Preliminary Results from Gerstein Lab on Structural Variation
The Gerstein lab has much experience in developing methods for detecting and characterizing structural variants based on the results of NextGen DNA sequencing and carrying out this work on a large-scale in the framework of the 1000 Genomes Project.
1 PEMer (Korbel et al. 2009)
We have developed a computational analysis pipeline Paired-End Mapper (PEMer) (Korbel 2007 & Korbel 2009) for finding SVs from paired-end 454/Roche sequencing data at high-resolution (see Figure 1). PEMer proceeds by optimally mapping paired-end reads to the reference human genome. Then, deletions and insertions relative to the reference genome are identified as regions with discordantly mapped paired-ends supported by multiple reads, i.e. those having too long or too short span compared to the average span. Moreover, comparing the relative orientations or positions of mapped ends can identify inversions or more complex SV events relative to the reference genome. We further extended PEMer to consider the number of supportive paired-ends, as well their length to call for an SV (Korbel 2009). Such a strategy allows utilizing more discordant (by relaxing cutoffs) paired-ends for SV calling and, eventually, leads to better sensitivity in finding SVs (especially SVs of small size) and enhancement of the resolution of the approach (by ~1.3 kb). With the aid of realistic simulation that considers various types of experimental errors, we optimized PEMer to yield maximum sensitivity while keeping the false positive rate at a desired level. For example, we found that at 8x physical coverage PEMer is able to find ~90% of heterozygous deletions (homozygous events are easier to detect) larger than 4 kb with false-positive rate of ~5%. Remarkably, PEMer demonstrated similar sensitivity (by detecting 88% of fosmid sequenced deletions) and false-positive rate (~6%, based on large scale PCR and aCGH validation) when assessed on 454 paired-end data in frames of 1000 genomes project.
PEMer has been applied to the 454 paired-end data (with ~2.5kb insert size) in pilot phase of the 1000 Genomes Project. The seven individuals in the dataset are from 3 CEPH families. The statistics of deletions, insertions and inversions detected for each of the individuals are displayed in Table 1. Particularly, NA12878, the daughter of a CEPH trio, yielded an effective coverage of ~8X of the diploid human genome and 1062 SVs (575 deletions, 296 insertions and 191 inversions) were identified from it. Further, 1585 additional SVs were identified from the other low coverage individuals.
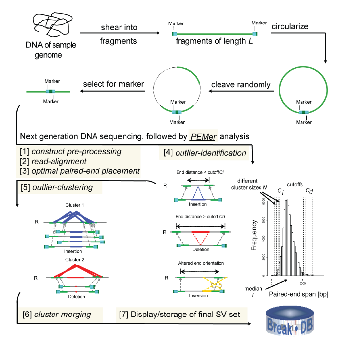
Fig 1 - PEMer workflow
|
Table 1. Statistics on preliminary SV calls with PEMer for 454 paired-end data from Freeze 1 and Freeze 2 1000 Genomes Project |
||||||||||||||||||||||||||||||||||||||||
|
2 MSB (Wang et al. 2008)
We have also developed a preliminary approach for read-depth (RD) analysis, MSB. It is an extension of our previous work on segmentation for array-based comparative genomic hybridization (aCGH) (Korbel et al., 2007 CGH, Wang et al., 2008). In general, most the previous approaches described for RD analysis attempt to explicitly model the underlying distribution of data based on particular assumptions. Often, they optimize likelihood functions for estimating model parameters, by expectation maximization or related approaches; however, this requires good parameter initialization through pre-specifying the number of segments. Moreover, convergence is difficult to achieve, since many parameters are required to characterize an experiment. To overcome these limitations, we proposed a nonparametric method without a global criterion to be optimized. This method involves mean-shift-based (MSB) procedures; it considers the observed RD signal as sampling from a probability-density function, uses a kernel-based approach to estimate local gradients for this function, and iteratively follows them to determine local modes of the signal. Overall, this method achieves robust discontinuity-preserving smoothing, thus accurately segmenting chromosomes into regions of duplication and deletion. It does not require the number of segments as input, nor does its convergence depend on this.
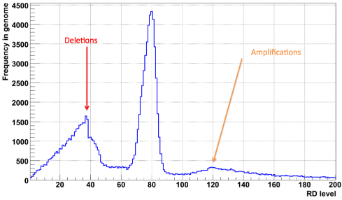
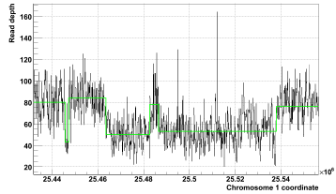
We have successfully applied the method to Solexa data in the 1000 Genomes Project. We see pronounced peaks in partitioned RD level distribution for homozygous deletions and amplifications (see Fig. 2). Example of successful partitioning in a difficult case is shown on Fig. 3. The overlap with validated SV regions suggests that our approach can detect >90% of RD-discoverable SVs (balanced SVs are not discoverable by RD).
|
|
|
Figure 2. Read depth level distribution after partitioning by MSB method. Arrows point peaks corresponding to heterozygous deletions and amplifications. |
|
|
|
Figure 3. Reconstruction of three deletions in the displayed regions of NA12878 individual is complicated by close proximity of the deletions, their largely different sizes and extreme signal noise. Partitioning by MSB approach (green line) allows for successful identification of all of them. |
3 ReSeqSim (Du et al. 2009)
Most recently, we have also built a simulation toolbox that will help optimize the combination of different technologies to perform comparative genome re-sequencing, especially in reconstructing large SVs (Du et al. 2009). In recent years, there has been great excitement in the development of many different technologies for this purpose (e.g. long, medium and short read sequencing from such as 454 and SOLiD, or high-density oligo-arrays with different probe spacing from Affymetrix and NimbelGen), with even more expected to appear. The costs and sensitivities of these technologies differ considerably from each other. As an important goal of personal genomics is to reduce the total cost of individual re-sequencing to an affordable point, it is worthwhile to consider optimally integrating technologies.
We have built a simulation toolbox that will help optimize the combination of different technologies to perform comparative genome re-sequencing, especially in reconstructing large SVs, which is considered in many respects the most challenging step in human genome re-sequencing, and is sometimes even harder than de novo assembly of a small genome because of the existence of duplications and repetitive sequences. To this end, we have formulated canonical problems that are representative of the issues in reconstruction, and which are of small enough scale to be computationally tractable and readily simulable. Using semi-realistic simulations, we are able to investigate how to combine different technologies to optimally solve the assembly at low cost. With mapability maps, our simulations efficiently take into account the inhomogeneous repeat-containing structure of the human genome and the computational complexity of practical assembly algorithms They quantitatively show combining different read lengths is more cost effective than using one length, how an optimal mixed sequencing strategy for reconstructing large novel SVs usually also gives accurate detection of SNPs and indels, how paired-end reads can improve the reconstruction efficiency, and how adding in arrays are more efficient than just sequencing for disentangling some complex SVs.
We envision that in the future, more experimental technologies can be incorporated into this sequencing/assembly simulation and the results of such simulations can provide informative guidelines for the actual experimental design to achieve optimal assembly performance at relatively low costs. With this purpose, we have made this simulation framework downloadable as a general toolbox that can be either used directly or extended easily.
4 Correlating SDs and CNVs (Kim et al. 2008)
We examined the formation signatures of CNV and SD and found that Alus, believed to be the major mediators of SD formation, have a decreased effect on younger SD and CNV formation, while non-homologous end joining (NHEJ) and repeats, such as LINE 1 elements and microsatellites, have become more prominent drivers of formation (Kim et al. 2008).
REFERENCES
Du J, Bjornson RD, Zhang ZD, Kong Y, Snyder M, Gerstein M (2009) Sequencing technology integration in personal genomics: optimal low cost reconstruction of large structural variants. PLoS Computational Biology, in press.
Kim, P.M. et al. Analysis of copy number variants and segmental duplications in the human genome: Evidence for a change in the process of formation in recent evolutionary history. Genome Res. 2008 Dec;18(12):1865-74.
Korbel, J. O., A. Abyzov, et al. (2009). "PEMer: a computational framework with simulation-based error models for inferring genomic structural variants from massive paired-end sequencing data." Genome Biol 10(2): R23.
Korbel, J. O., A. E. Urban, et al. (2007). "Paired-end mapping reveals extensive structural variation in the human genome." Science 318(5849): 420-6.
Wang, LY, A Abyzov, JO Korbel, M Snyder, M Gerstein (2009) MSB: a mean-shift-based approach for the analysis of structural variation in the genome. Genome Res 19: 106-17.
3