C. Progress Report & Preliminary Studies
Progress on Surveys of Membrane Protein Folds and Families in Genomes
During the preceding grant period, we completed a number of surveys of membrane
proteins in genomes.
Occurrence in Gene Families: Gerstein et al. (2000), Liu et al. (2002,
2003), Lehnert et al. (2004)
In Gerstein et al. (2000), we surveyed the occurrence of membrane proteins in
the worm genome. This highlighted the large number of 7-TM proteins in this
organism. In Liu et al. (2002, 2003) and Lehnert et al., (2004) we classified
polytopic membrane protein domains on the basis of sequence similarities and
topology using existing PFAM families. Using the classified families, we applied
a genome-wide analysis on the occurrence of the families and on the patterns
of conserved amino acids and motifs in the TM-helix regions in the families.
Some interesting trends identified include: (i) There is an approximately linear
relationship between the number of classified membrane protein domains and the
number of open reading frames (ORFs). (ii) The majority of integral membrane
proteins have only a single polytopic membrane domain, suggesting recombination
of domains is not common inside membranes. Distinct from soluble proteins, which
gain new functions by recombination of different domains in the course of evolution,
membrane proteins could achieve the same goal by non-covalent oligomeric associations
within the membrane.
Occurrence in Pseudogene Families: Zhang et al. (2003) and Zhang &
Gerstein (2003)
We have extended the work on classifying membrane proteins into families and
folds to encompass pseudogenes. Pseudogenes are disabled copies of functional
genes in the genome; these sequences have close similarities to one or more
paralogous functional genes, but in general are unable to be transcribed (Vanin
1985; Mighell et al. 2000) . Our recent whole-genome survey has identified more
than 10,000 pseudogenes in the human genome and about 5,000 pseudogenes in the
mouse genome (Zhang et al., 2003). In the course of this overall survey we identified
many pseudogenes associated with membrane proteins and membrane-associated proteins
that we classified into families. A good example is cytochrome b (cytb), which
is a ubiquitous 8-TM protein that catalyzes a crucial step in the mitochondrial
oxidative phosphorylation process (Zhang et al. 1998; Zhang et al. 2000). The
functional gene of this protein is in the mitochondrial genome, but more than
70 copies of its cytb pseudogenes are present in the nuclear genome due to a
DNA-mediated process (Tourmen et al. 2002; Woischnik and Moraes 2002). We also
found that Cytochrome c (cyc), an important protein in the mitochondrial electron-transfer
chain that interacts with cytochrome b has 49 pseudogenes in the human genome
(Zhang & Gerstein, 2003).
Progress on Analysis of Protein Motifs, particularly in Membranes and Helices
Senes et al. (2000). In a collaborative project with the Engelman lab, we were able to comprehensively survey the occurrence of residue pairs and triplets in transmembrane helices. This study highlighted the commonness of the GxxxG motif, which had been previously identified in the experimental screens (Russ & Engelman, 2000). These statistical results about the commonness of particular motifs in membrane proteins have proven useful in membrane protein design -- i.e. in designing dimerization motifs.
Das & Gerstein (2000) and Schneider et al., (2002). In these two papers,
we did composition and motif analysis of thermophilic proteins, focusing on
the determinants of thermostability. Das et al. (2000) focused on the occurrence
of salt bridges in helices, and Schneider et al. (2002) focusing on the implications
of composition for membrane-protein helix-helix interactions.
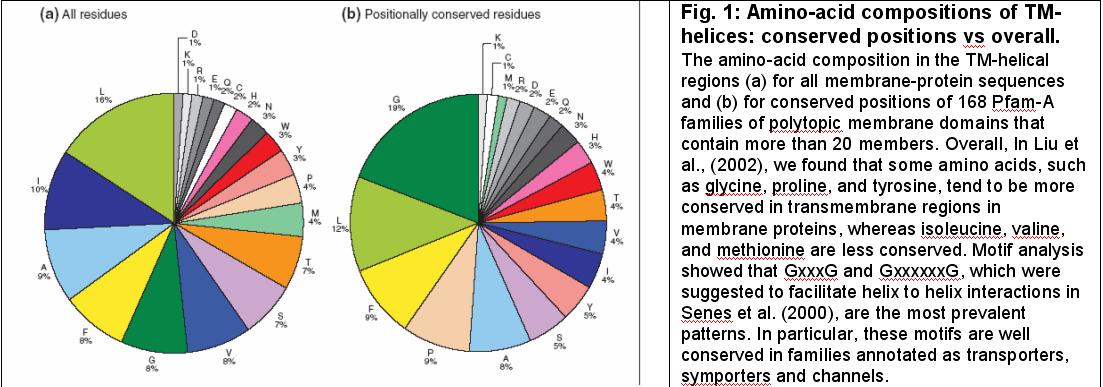
Liu et al. (2002) and Zhang et al., (2002b). As shown in Fig 1, in Liu et al. (2002), we found certain amino acids and motifs were more conserved in membrane proteins. In Zhang et al. (2002b), we extended our motif work further, looking at the occurrence of protein motifs, not only in known proteins but also in the intergenic regions of genome. We called the motifs found "pseudomotifs" (in the spirit of pseudogenes). We compared a few eukaryotic genomes in terms of occurrence of these motifs.
Freeman-Cook et al. (2004). We have developed an approach in close collaboration
with Prof. DiMaio to identify consensus sequences that promote interaction between
specific TM helices associated with the BPV E5 protein and the PDGF? receptor.
So far we have found that only a subset of all residues of the E5 transmembrane
domain is important for interaction with the PDGF? receptor (including L18,
V24, L25, V28, V20, L21, V23, and L29). An optimal consensus sequence with only
6 out of 15 variable positions specified can correctly classify 84% of all clones
as either transforming or non-transforming. If more parameters are allowed to
vary (as in the case of a scoring matrix), all but 2 clones can be correctly
classified. These optimal consensus sequences and scoring matrices can also
guide the design of new sequences. Finally, we showed that transforming clones
and non-transforming clones can be effectively differentiated computationally,
if and only if L and V are grouped into separate categories.
Progress on Helix Packing Calculations
Preliminary Membrane Protein Calculations: Gerstein & Chothia (1999)
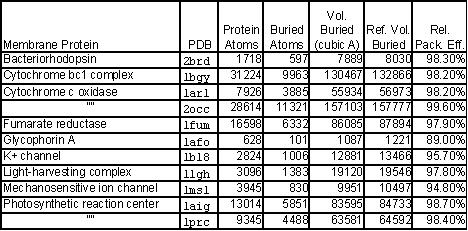
Early in the term of the grant, we did preliminary calculations on the packing
in membrane proteins. Our results, which are shown in the table to the right
and at molmovdb.org/ geometry/membrane suggest that membrane proteins are packed
more tightly than soluble ones. It is worth mentioning that simultaneous to
this, others worked on this problem (Eilers et al. 2000). They also demonstrated
that transmembrane proteins are more tightly packed, on average, then their
soluble counterparts despite the fact that transmembrane proteins cannot make
use of hydrophobic effects in folding within the bilayer. In our estimation,
there were two major problems with these early calculations: (1) there were
few membrane protein structures available at the time, and (2) we lacked a proper
packing parameter set. The first point is addressed simply by the passage of
time and the solving of more structures. We set about addressing the second
point in subsequent studies.
Packing Parameters: Tsai et al. (2001), Gerstein & Richards (2001) and
Tsai & Gerstein (2002)
We have been developing the methodology of packing calculations considerably.
Before they can be applied accurately to helix-helix interfaces, one needs a
consistent set of VDW parameters and standard volumes. This is presented in
the first paper, where we develop the ProtOr parameter set for packing calculations.
The second paper presents a comprehensive survey of packing calculations, placing
the new ProtOr set in context. In a third paper, we performed a detailed sensitivity
analysis of our calculations and created a database of relevant parameters,
which is available through the web. This sensitivity analysis was invaluable
in helping us understand which parameters were most useful in our packing calculations.

Progress on Development of Integrative Database Systems
Qian et al. (2001a) and Lin et al. (2002). The last period of this grant partially funded the development of two interlinked and integrated database systems, PartList.org and GeneCensus.org (described in Fig 2). In general, GeneCensus takes a more sequence and less structural view of genome comparisons than PartsList, focusing on expression data, pathway activities, and protein interactions.
Cheung et al. (2001), Greenbaum et al. (2001), Bertone & Gerstein, (2001). We have done a number of conceptual papers on issues in database integration. We have developed a number of approaches that work towards the goal of an integrated genomic database. The first paper above describes ways of using XML to allow the interoperation of separate genomic resources. The second two papers describe how we can think of genomic information as a succession of different "omes" that can be interrelated in various ways. These papers are conceptual but are valuable in the way that they help structure information and enable it to be laid out in coherent database framework.
Yu et al. (2004). Real protein networks are quite complex and can often be divided into many sub-networks through systematic selection of different nodes and edges. For instance, proteins can be sub-divided by expression level, length, amino-acid composition, solubility, secondary structure and function. A challenging research question is to compare the topologies of sub-networks, looking for global differences associated with different types of proteins. We recently created TopNet, an automated web tool designed to address this question (http://networks.gersteinlab.org/genome/interactions/networks), calculating and comparing topological characteristics for different sub-networks derived from any given protein network.
Progress Interrelating Membrane Proteins with Functional Genomics Data
We have used database integration to find a number of interesting correlations related to membrane proteins.
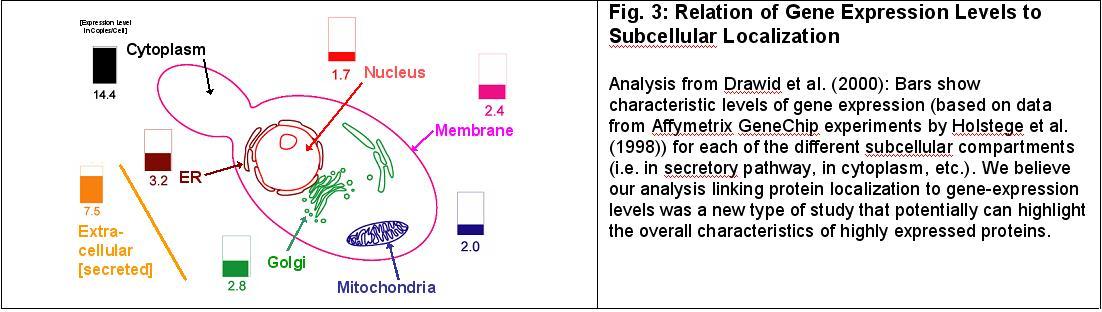
Jansen & Gerstein (2000) and Drawid et al. (2000). Using our integrated database system, in the first paper we were able to connect the prediction of transmembrane helices in yeast with a number of datasets giving measurements of whole genome expression levels. This produced the notable result that membrane proteins are expressed at a considerably lower level than soluble proteins, by ~22%, and that certain broad groups of membrane proteins are expressed more highly than others -- e.g. 4-TMs are expressed at a higher level than 2-TMs. In the second paper (described in Fig. 3), we extended this analysis to fully relate subcellular localization with gene expression level.
Drawid & Gerstein (2000), Alexandrov & Gerstein, (2001), Kumar et al. (2002). In the first paper we extended our work on gene expression and subcellular localization to the prediction of subcellular localization based on genomic features. That is, we made use of the correlations that we had found and used them for the overall prediction of subcellular localization. In the second paper, we developed the mathematics of our formalism further, showing that it was in many respects analogous to the probabilistic calculations in quantum mechanics. Finally, in the third paper, we scaled our procedure up and applied it to the whole yeast genome in combination with experiment, to predict subcellular localization for all proteins in yeast. This paper represents an experimental capstone on our computational work in this area. It will be broadly useful to yeast biologists.