Gerstein
Intergenic Annotation. The Gerstein lab has been a major
participant in a number of ENCODE efforts, focusing on intergenic annotation.
We developed an approach to analyze the distribution of regulatory elements
found in many different ChIP-chip experiments (Zhang et al., 2006). We focused
on the overall chromosomal distribution of regulatory elements in the encode
regions and showed that it is highly nonrandom. Our results indicate that these
elements are clustered into regulatory rich ‘islands’ and poor ‘deserts.’ We then
performed a multivariate analysis on all the factors collectively. This grouped
the transcription factors into sequence-specific and sequence-nonspecific
clusters. Following up on this, we
developed an approach for integrating the results of many ChIP-chip experiments
to discover new promotors in the human ENCODE regions (Trinklein et al. 2006).
We also have carried out comprehensive pseudogene annotation of the human
genome and cross referenced this annotation with tiling arrays (Zheng et al.,
2005, 2007; Harrison et al., 2005; Zheng et al., 2006; Zheng & Gerstein,
2006, 2007; Zhang et al., 2003). We performed a related analysis of structured
RNAs in intergenic regions, also inter-relating them with tiling array data
(Zhang et al., 2007; Washietl et al., 2007).
Tiling Array Tools. The Gerstein Lab has developed a
considerable amount of tools and machinery for processing tiling arrays. Most
of these are described elsewhere in the proposal (e.g. scoring arrays, sect. C.4).
One tool not described elsewhere is BoCaTFBS (Wang et al., 2006). This refines
ChIP-chip hits by considering known motifs (Wang et al., 2006). Traditional
computational algorithms used to identify binding sites, such as consensus
sequences (Osada et al., 2004), profile methods (PWM/PSSM) (Stormo, 2000), and
HMMs (Pavlidis et al., 2001; Ellrott et al., 2002) generate high false-positive
rates when applied genome-wide (Wasserman & Sandelin, 2004). Our method
uses a boosted cascade of classifiers -- specifically, alternating decision
trees (ADTboost) (Freud & Schapire, 1999), where ADTboost is a special
extension of AdaBoost. We use the known motifs (e.g. from the TRANSFAC
database, Matys et al., 2003) as positives and the results of the ChIP-chip
experiments as negatives. Our method is the first motif finder that explicitly
takes into account the data from ChIP-chip experiments. Moreover, BoCaTFBS
differs from most other motif programs in that (1) it takes into consideration
positional dependencies within a given motif and (2) it uses the negative data
(regions where the transcription factor does not bind) in order to refine the
binding site.
Regulatory Networks. The Gerstein lab has done quite a
number of analyses on the large-scale structure of regulatory networks (e.g. Yu
& Gerstein, 2006; Luscombe et al., 2004; Yu et al., 2003, 2004) and has
developed tools to enable their analysis (Yip et al., 2006; Yu et al., 2004;
networks.gersteinlab.org/tyna ).
Publications
* Zhengdong, Z., Alberto, P., Yutao F., Sherman W., Zhiping
W., Chang J., Snyder M, Gerstein M. Development of approaches for global
analysis of regulatory elements. Global analysis of the genomic distribution
and correlation of regulatory elements in the Encode regions. Genome Research. Genome
Res. In press.
* Euskirchen, G., Rozowsky, J., Wei, C.L. Lee, W.H., Zhang,
Z, Hartman, S., Emanuelsson, O., Stolc, V., Weissman, S., Gerstein, M., Ruan,
Y., Snyder, M. (2006). Mapping of Transcription
Factor Binding Regions in Mammalian Cells by ChIP: Comparison of Array
and Sequencing Based Technologies. Genome Res. In press.
* Trinklein ND, Karaöz U, Wu J, Halees A, Force
Aldre S, Collins PJ, Zheng D, Zhang Z, Gerstein M, Snyder M, Myers RM, Weng, Z.
Integrated analysis of experimental datasets reveals many novel promoters in 1%
of the human genome. Genome
Research., in press.
* S Washietl, JS Pedersen, JO Korbel, AR Gruber,
J Hackermuller, J Hertel, M Lindemeyer, K Reiche, C Stocsits, A Tanzer, C Ucla,
C Wyss, SE Antonarakis, F Denoeud, J Lagarde, J Drenkow, P Kapranov, TR
Gingeras, M Snyder, MB Gerstein, A Reymond, IL Hofacker, PF Stadler .
Structured RNAs in the ENCODE Selected Regions of the Human Genome Genome
Research (in press)
* J Du, JS Rozowsky, JO Korbel, ZD Zhang, TE
Royce, MH Schultz, M Snyder, M Gerstein (2006) A supervised hidden markov model
framework for efficiently segmenting tiling array data in transcriptional and
chIP-chip experiments: systematically incorporating validated biological
knowledge.Bioinformatics 22: 3016-24.
* Zhang Z, Rozowsky J, Lam HYK, Snyder M,
Gerstein M. Tilescope: online analysis pipeline for high-density tiling
microarray data GenomeBiology (in press).
* Wang L, Comaniciu D, Snyder M, Gerstein M.
BoCaTFBS: a boosted-cascade learner to refine the binding sites suggested by
ChIP-chip experiments. GenomeBiology. Genome Biol 7: R102
Bertone P, Trifonov V, Rozowsky JS, Schubert F, Emanuelsson
O, Karro J, Kao MY, Snyder M, Gerstein M. (2006) Design optimization
methods for genomic DNA tiling arrays. Genome Res. 16: 271-81.
Borneman,
AR, Leigh-Bell, J, Yu, H, Bertone, P., Gerstein, M., Snyder,.M (2006) Target
Hub Proteins Serve as Master Regulators of the Complex Transcriptional Network
Controlling Yeast Pseudohyphal Growth. Genes Dev 20: 435-448.
* Rozowsky J, Newburger D, Sayward G, Wu J,
Jordan G, Korbel JO, Nagalakshmi U, Yang J, Zheng D, Guigo R, Gingeras T,
Weissman S, Miller P, Snyder M,
Gerstein M. The DART Classification of Unannotated Transcription within the ENCODE
Regions: Associating Transcription with Known and Novel Loci Genome Research
(in press)
Thomas E. Royce, Joel S. Rozowsky and Mark B.
Gerstein (2007). Assessing the need for sequence-based normalization in tiling
microarray experiments. Bioinformatics (in press).
* Zheng D, Gerstein M. A computational approach
for identifying pseudogenes in the ENCODE regions. Genome Biol 7 Suppl 1: S13.1-10.
* Emanuelsson O, Nagalakshmi U, Zheng D, Rozowsky JS, Urban
AE, Du J, Lian Z, Stolc V, Weissman S, Snyder M, Gerstein M. (2006) Assessing the performance
of different high-density tiling microarray strategies for mapping transcribed
regions of the human genome.
Genome Research, in press.
Royce, T.E., Rozowsky, J.S., Bertone, P., Samantac, M.,
Stolc, V., Weissman, S., Snyder, M. and Gerstein, M. (2005). Issues in the
Analysis of Oligonucleotide Tiling Microarrays. Trend Genetics 21: 466-475.
H Yu, M Gerstein (2006) Genomic analysis of the hierarchical
structure of regulatory networks. Proc Natl Acad Sci U S A 103: 14724-31.
C4.2.
Analysis of the ChIP samples
C4.2a. Comparison of
ChIP-chip Platforms and Parameters. At the
outset of the ENCODE pilot phase, both PCR product arrays and high density oligonucleotide
(HDO) arrays were being used by various groups. However, the overall
performance and suitability for genome-scale analyses had not ever been
compared. Therefore, we began by comparing PCR arrays and 390,000 feature
arrays prepared by maskless photolithography (e.g. NimbleGen) by performing
ChIP-chip for three yeast factors (Borneman et al., 2006b). We found that the
HDO arrays detected approximately three times more binding events than the PCR
arrays while also showing increased accuracy. We also investigated optimal
parameters for mapping binding sites in mammalian cells using ChIP-chip
(Euskirchen et al., 2007). We
compared parameters such as DNA microarray format (oligonucleotide versus PCR),
oligonucleotide length, hybridization conditions, and the use of competitor Cot
DNA. Also, methods were devised
for scoring and comparing results.
Optimal signal-to-noise, sensitivity and specificity was observed with
HDO arrays relative to PCR product arrays (Figure 2). We observed optimal signals using oligonucleotides >36 b
and the presence of Cot competitor DNA (See Figure 2 for the oligonucleotide
length results). Consistent with
the better performance of arrays containing longer oligonucleotides, we were
not able to identify STAT1 targets using Affymetrix arrays (which uses 25
mers). Target identification as a
function of biological replicates was also determined and revealed that 80-86%
of targets were identified with three experimental repeats; four provides a
modest 3-5% increase (Euskirchen et al., 2007). We also did a
parallel study as the part of the ENCODE consortium to compare platforms for
the suitability for assaying transcription (Emanuelsson et al., 2006). Overall, our conclusions were similar
to those in Euskirchen et al. (2007) -- that is, HDO arrays performed better than PCR ones and
feature density was all important.
However, for transcript mapping we got better performance with 25mers
than 36mers. Thus, the HDO array platform provides a far more robust array
system by all measures than PCR-based arrays, all of which is directly
attributable to the large number of probes available. Since we performed this
study Agilent has also begun to manufacture HDO arrays. However their highest density array
(244K features) and price ($500) does not make them competitive with the
Nimblegen whole genome 10 array set (2.1 million features/array). Affymetrix
arrays are competitive in price ($1500 for one set). However, some of our team
members have found that they are not as effective as NimbleGen arrays when
analyzing certain factors and they cannot be reused (Nimblegen arrays can be
used at least twice and new chemistry that should increase the number of
rehybridizations/array is currently being tested by one of our team members in
collaboration with NimbleGen).
Thus, at this time Nimblegen arrays provide the highest sensitivity and
the most value of the array platforms
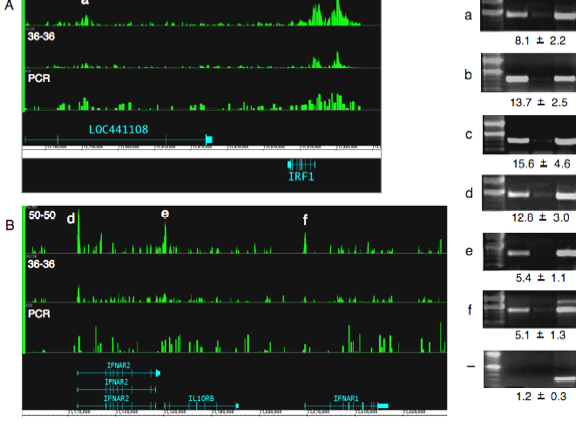
Figure 2 Comparison of
ChIP-chip results using different array platforms and oligonucleotide lengths. Raw signals from each oligonucleotide are plotted. Key: PCR: PCR product array. 36-36 36 b oligonucledotides arrayed
end to end; 50-50: 50 b oligonucledotides arrayed end to end; a-f validated
positive regions - negative control region.
C4.2b Comparison of ChIP-chip with ChIP-Sequencing. We have also compared ChIP-chip to
ChIP followed by DNA sequence analysis of fragment ends (ChIP-PET; Ng et al.,
2006) for the STAT1 transcription factor (Euskirchen et al., 2007). We found that these two technologies
were comparable in terms of sensitivity and specificity (Figure 3). Seven of 8
targets enriched 4-fold or more were found with both methods; 4 fold is
approximately the threshold at which ChIP targets reproducibly validate by qPCR
(see below). The new 2.1 million feature array design contains more probes in
repeated regions and is expected to identify all 8 (Euskirchen et al 2006). The
resolution of ChIP chip is generally higher (targets can be mapped and
validation to within 200 bp (K.S., P.R., M. S., unpublished; see also XXX) than
ChIP PET, particularly on the less enriched peaks (see figure 3). At this
point, ChIP-PET is also considerably more expensive, even with the new 454
sequencing technologies, than analysis of samples using the NimbleGen 10 array
set. However, we will continue to compare ChIP-chip and ChIP-sequencing during
the production phase and always make sure that we are using the technique that
provides the highest quality and most comprehensive data for the lowest price.
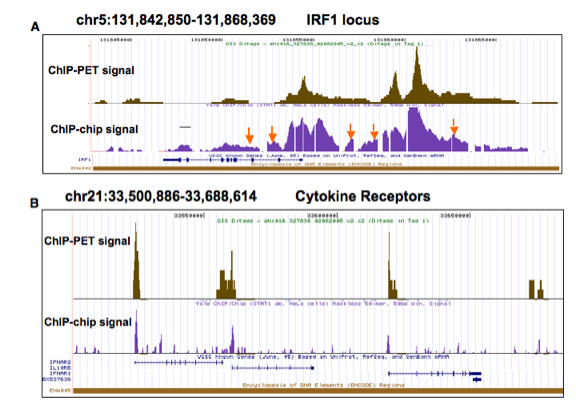
Figure 3. Comparison
of ChIPchip and ChIP-PET results. Raw signals from each method
are plotted. The concordance of
the results for both signals (shown above) and called target regions is quite
high (greater than 90% for the top targets (estimated to be 4 fold enriched).
C5. Bioinformatics - Processing pipeline
for target identification and integration of primary data (ChIP-chip) with
secondary validation data.
C5.1. Array Processing Platforms
-- ExpressYourself & Tilescope
We have developed two generations of tiling arrays software: ExpressYourself for PCR arrays (Luscombe et al., 2003; bioinfo.mbb.yale.edu/ExpressYourself) and Tilescope for oligo arrays (Zhang et al., 2007; tilescope.gersteinlab.org). Tilescope is an automated data processing pipeline for analyzing data sets generated in experiments using high-density tiling microarrays. The software performs data normalization, combination of replicate experiments, tile scoring, and feature identification. Given the modular architecture of the pipeline, new analysis algorithms can be readily incorporated. Tilescope is capable of handling very large data sets, such as ones generated by whole genome ChIP-chip experiments, as we developed an efficient new data compression algorithm to reduce the data size for fast online data transmission. Tilescope is designed with a graphical user-friendly interface to facilitate a user’s data analysis task, and the results, presented on a web page, can be downloaded for further analysis.
C5.2. Approaches for Array
Scoring and Artifact Correction
Tilescope
incorporates a number of scoring and artifact correction approaches we have developed
for tiling arrays. In particular, for Nimblegen arrays, we identified those
procedures in the microarray normalization literature that are transferable to
tiling arrays and those that needed to be modified or replaced (Royce et al.,
2005, 2006). One issue is that a small fraction of probes on an array
experience significant levels of nucleic acid hybridization; many normalization
procedures assume hybridization to at least half of all probes on an
array. Another issue is that tiling arrays for whole genomes, for
example, require many arrays - each of different design. Care must be taken
when normalizing these arrays to one another because the underlying
distribution of measured signals may vary widely among arrays tiling different
regions of the genome. We have also worked extensively on compensating for
issues of cross-hybridization. In Royce et al. (2007) we developed a correction
system for non-specific, sequence-based cross hybridization that is readily
applicable to tiling arrays. We have also developed tools for the optimal
design of tiling arrays to minimize the effect of repetitive and specifically
cross-hybridizing probes (Bertone et al., 2006). These algorithms are
implemented as a collection of web-based tools, available at
tiling.gersteinlab.org. Finally, in processing large
amounts of expression data it is important to be able to discover and correct
various spatial array artifacts. Qian et al. (2003), Kluger et al.
(2003), and Yu et al., (2007) developed a way of measuring and quantifying a
common spatial artifact that can induce spurious correlation of nearby spots on
the array.
C5.3. Tools to integrate the list of functional elements --
DART
We
have developed DART (DART.gersteinlab.org, Rozowsky et al., 2007) to facilitate
the flexible storage, visualization, and comparison of the growing number of
experimentally defined sets of transcription factor binding sites. DART has
been designed to address a number of challenging issues that arise when
attempting to analyze, compare, and store these types of data. The key aspects of DART include the
following: Dealing with heterogeneous datasets, flexibility for storing
different tiling array hit attributes, accommodating new genome builds, and integrated
linking to other web resources for broader visualization and analysis. Furthermore,
DART provides machinery for helping to pick targets for validation.
H. Literature Cited
Babu, M.M., Luscombe, N. M., Aravind, L., Gerstein, M. and Teichmann S.
A. (2004) Structure and evolution of transcriptional regulatory networks. Curr
Opin Struct Biol, 14, 283-91.
Bailey, T.L. and Elkan, C. (1994). Fitting a mixture model by
expectation maximization to discover motifs in biopolymers. Proc Int Conf
Intell Syst Mol Biol 2: 28-36.
Bailey, T.L., Williams, N., Misleh, C. and Li, W.W. (2006). Links MEME: discovering and analyzing DNA and protein sequence motifs. Nucleic
Acids Res: W369-73.
Bertone, P., Kluger, Y., Lan, N., Zheng, D., Christendat, D., Yee, A.,
Edwards, A. M., Arrowsmith, C. H., Montelione, G. T.and Gerstein, M. (2001)
SPINE: an integrated tracking database and data mining approach for identifying
feasible targets in high-throughput structural proteomics. Nucleic Acids Res,
29, 2884-98.
Bertone, P., Stolc, V., Royce, T.E., Rozowsky, J.S., Urban, A.E., Zhu,
X., Rinn, J.L.,Tongprasit, W., Samanta, M., Weissman, S., Gerstein, M., and
Snyder, M. (2004). Global Identification of Human Transcribed Sequences with
Genome Tiling Arrays. Science 306: 2242-6.
Bertone, P., Trifonov, V., Rozowsky, J.S., Schubert, F., Emanuelsson,
O., Karro, J., Kao, M.Y., Snyder, M., Gerstein, M. (2006) Design optimization
methods for genomic DNA tiling arrays. Genome Res 16: 271-81.
Birney, E., Andrews, D., Caccamo, M., Chen, Y., Clarke, L., Coates, G.,
Cox, T., Cunningham, F., Curwen, V., Cutts, T., Down, T., Durbin, R.,
Fernandez-Suarez, X.M., Flicek, P., Graf, S., Hammond, M., Herrero, J., Howe,
K., Iyer, V., Jekosch, K., Kahari, A., Kasprzyk, A., Keefe, D., Kokocinski, F.,
Kulesha, E., London, D., Longden, I., Melsopp, C., Meidl, P., Overduin, B.,
Parker, A., Proctor, G., Prlic, A., Rae, M., Rios, D., Redmond, S., Schuster,
M., Sealy, I., Searle, S., Severin, J., Slater, G., Smedley, D., Smith, J.,
Stabenau, A., Stalker, J., Trevanion, S., Ureta-Vidal, A., Vogel, J., White,
S., Woodwark, C. and Hubbard, T.J. (2006). Ensembl 2006. Nucleic Acids Res: D556-61.
Bolstad, B.M., Irizarry, R.A., Astrand, M. and Speed, T.P. (2003).
Summaries of Affymetrix GeneChip probe level data. Bioinformatics 19: 185-193.
Borneman, A.R., Leigh-Bell, J., Yu, H., Bertone, P., Gerstein, M., and
M. Snyder. (2006) Target Hub Proteins Serve as Master Regulators of the Complex
Transcriptional Network Controlling Yeast Pseudohyphal Growth. Genes Dev 20:
435-448.
Boyer, L. A., Lee, T. I., Cole, M. F., Johnstone, S. E., Levine, S. S.,
Zucker, J. P., Guenther, M. G., Kumar, R. M., Murray, H. L., Jenner, R. G., et
al. (2005). Core transcriptional regulatory circuitry in human embryonic stem
cells. Cell 122: 947-956.
Buchholz, F., Angrand, P. O., and
Stewart, A. F. (1996). A
simple assay to determine the functionality of Cre or FLP recombination targets
in genomic manipulation constructs. Nucleic Acids Res 24: 3118-3119.
Buchholz, F., Angrand, P. O., and Stewart, A. F. (1998). Improved
properties of FLP recombinase evolved by cycling mutagenesis. Nat Biotechnol
16: 657-662.
Carriero, N., Osier, M. V., Cheung, K. H., Miller, P. L., Gerstein, M.,
Zhao, H., Wu, B., Rifkin, S., Chang, J., Zhang, H., White, K., Williams, K. and
Schultz, M. (2005)A high productivity/low maintenance approach to
high-performance computation for biomedicine: four case studies. J Am Med
Inform Assoc 12: 90-8.
Cawley, S., Bekiranov, S., Ng, H.H., Kapranov, P., Sekinger, E.A.,
Kampa, D., Piccolboni, A., Sementchenko, V., Cheng, J., Williams, A.J.,
Wheeler, R., Wong, B., Drenkow, J., Yamanaka, M., Patel, S., Brubaker, S.,
Tammana, H., Helt, G., Struhl, K., Gingeras, T.R. (2004). Unbiased mapping of
transcription factor binding sites along human chromosomes 21 and 22 points to
widespread regulation of noncoding RNAs. Cell 116: 499-509.
Chen, Q., Hertz, G. and Stormo, G. (1995). Matrix Search 1.0: A computer
program that scans DNA sequences
for transcriptional elements using a database of weight matrices. Comput Appl
Biosci, 11: 563-566.
Cheung K.H., White, K., Hager, J., Gerstein, M., Reinke, V., Nelson, K.,
Masiar, P., Srivastava, R., Li, Y., Li, J., Zhao, H., Li, J., Allison, D.B.,
Snyder, M., Miller, P., Williams, K. (2002). YMD: A microarray database for
large-scale gene expression analysis. Proc. AMIA Symp: 140-144.
Cheung, K. H., Smith, A. K., Yip, K. Y. L., Baker, C. J. O. and Gerstein,
M. (2007). Semantic Web Approach to Database Integration in the Life Sciences
in Semantic Web: Revolutionizing Knowledge Discovery in the Life Sciences (eds.
C Baker and K Cheung, Springer, NY), pp. 11-30.
Cheung, K. H., Yip, K. Y., Smith, A., Deknikker, R., Masiar, A. and
Gerstein, M. (2005) YeastHub: a semantic web use case for integrating data in
the life sciences domain. Bioinformatics, 21, Suppl 1, i85-96.
Conlon, E.M., Liu, X.S., Lieb, J.D., Liu, J.S. (2003). Integrating
regulatory motif discovery and genome-wide expression analysis. Proceedings of
the National Academy of Sciences USA 100: 3339-3344.
Copeland, N. G., Jenkins, N. A., and Court, D. L. (2001).
Recombineering: A powerful new tool for mouse functional genomics. Nat Rev
Genet 2: 769-779. Database. J Am Med Inform Assoc 1998 Nov-Dec 5(6): 511-27.
Deplancke, B., Dupuy, D., Vidal, M., and Walhout, A.J. (2004). A gateway-compatible yeast one-hybrid
system. Genome Res. 14: 2093-101.
Du, J., Rozowsky, J.S., Korbel. J., Zhang, Z., Royce, T.E., Schultz,
M.H., Snyder, M., and Gerstein, M. (2006). A supervised hidden markov model
framework for efficiently segmenting tiling array data in transcriptional and
chip-chip experiments systematically incorporating validated biological
knowledge. Genome Res. Submitted.
Ellrott, K., Yang, C., Sladek, F.M., Jiang, T. (2002). Identifying
transcription factor binding sites through Markov chain optimization.
Bioinformatics, 18 suppl 2: S100-S109.
Emanuelsson, O., Nagalakshmi, U., Zheng, D., Rozowsky, J.S., Urban,
A.E., Du, J., Lian, Z., Stolc, V., Weissman, S., Snyder, M., and Gerstein, M.
(2006). Assessing the performance of different high-density tiling microarray
strategies for mapping transcribed regions of the human genome. Genome
Research. In press.
Euskirchen, G., Royce, T.E., Bertone, P., Martone, R., Rinn, J.L.,
Nelson, F.K., Sayward, F., Luscombe, N.M., Miller, P., Gerstein, M., (2004).
CREB binds to multiple loci on chromosome 22. Mol. Cell Biol. 24: 3804-3814.
Euskirchen, G., Rozowsky, J., Wei, C.L, Lee, W.H., Zhang, Z.,
Hartman,S., Emanuelsson, O., Stolc, V., Weissman, S., Gerstein, M., Ruan, Y.,
Snyder, M. (2006). Optimal Mapping of Transcription Factor Binding Sites in
Mammalian Cells. Genome Research. In press.
Feng, W., Wang, G., Zeeberg, B.R., Guo, K., Fojo, A.T., Kane, D.W.,
Reinhold, W.C., Lababidi, S., Weinstein, J.N., Wang, M.D. (2003). Development
of gene ontology tool for biological interpretation of genomic and proteomic
data. AMIA Annu Symp Proc.
Frith, M.C., Hansen, U., Spouge, J.L. and Weng, Z. (2004). Finding
functional sequence elements by multiple local alignment Nucleic Acids Res. 32:
189-200.
Frith, M.C., Li, M.C., and Weng, Z. (2003). Cluster-Buster: Finding
dense clusters of motifs in DNA sequences. Nuc Acids Res 13: 3666-3668.
Gabriel, K. R. (1971). The biplot graphical display of matrices with
application to principal component analysis. Biometrika 58: 453-467.
Gaudet, J., Muttumu, S., Horner, M., and Mango, S. E. (2004).
Whole-genome analysis of temporal gene expression during foregut development.
PLoS Biol 2: e352.
Gerstein, M. (2000) Annotation of the human genome. Science, 288,1590.
Gerstein, M. (2006) Tools needed to navigate landscape of the genome.
Nature, 440, 740.
Gerstein, M., Sonnhammer, E. L., and Chothia, C. (1994). Volume changes
in protein evolution. J Mol Biol 236: 1067-1078.
Giepmans, B. N., Adams, S. R., Ellisman, M. H., and Tsien, R. Y. (2006).
The fluorescent toolbox for assessing protein location and function. Science
312: 217-224.
Goh, C. S., Lan, N., Echols, N., Douglas, S. M., Milburn, D., Bertone,
P., Xiao, R., Ma, L. C., Zheng, D., Wunderlich, Z., Acton, T., Montelione, G.
T. and Gerstein, M. (2003) SPINE 2: a system for collaborative structural
proteomics within a federated database framework. Nucleic Acids Res, 31,
2833-8.
Greenbaum, D., Douglas, S.M., Smith, A., Lim, J., Fischer, M., Schultz,
M. and Gerstein, M. (2004) Computer security in academia-a potential roadblock
to distributed annotation of the human genome. Nat Biotechnol, 22, 771-2.
Gupta, M. and Liu, J.S. (2005). De novo cis-regulatory module
elicitation for eukaryotic genomes. Proc Natl Acad Sci U S A. 102: 7079-84.
Harbison, C.T., Gordon, D.B., Lee, T.I., Rinaldi, N.J., Macisaac, K.D.,
Danford, T.W., Hannett, N.M., Tagne, J.B., Reynolds, D.B., Yoo, J., Jennings,
E.G., Zeitlinger, J., Pokholok, D.K., Kellis, M., Rolfe, P.A., Takusagawa,
K.T., Lander, E.S., Gifford, D.K., Fraenkel, E., Young, R.A. (2004).
Transcriptional regulatory code of a eukaryotic genome. Nature 431: 99-104.
Harrison, P.M., Zheng, D., Zhang, Z., Carriero, N., and Gerstein, M.
(2005). Transcribed processed pseudogenes in the human genome: An intermediate
form of expressed retrosequence lacking protein-coding ability. Nucleic Acids
Res 33: 2374-83.
Hartman, S.E., Bertone, P., Nath, A., Royce, T.E., Gerstein, M.,
Weissman, S. and M. Snyder. (2005). Global Changes in STAT Target Selection and
Transcription Regulation Upon Interferon Treatments. Genes Dev 19: 2953-2968.
Horak, C. and Snyder, M. (2002). ChIP chip: A genomic Approach for
Identifying Transcription Factor Binding Sites. Methods Enzymol. 350: 469-484.
Horak, C.E., Luscombe, N.M., Qian, J., Piccirrillo, S., Gerstein, M, and
Snyder, M. (2002) Complex Transcriptional Circuitry at the G1/S Transition in
Saccharomyces cerevisiae. Genes Dev 16: 3017-3033.
Horak, C.E., Mahajan, M.C., Luscombe, N.M., Gerstein, M., Weissman,
S.M., and Snyder, M. (2002). GATA-1 binding sites mapped in the globin locus by
using Mammalian ChIp-chip analysis. Proc. Natl. Acad. Sci. U S A 99: 2924-2929.
Hudson, J.R. Jr., Dawson, E.P., Rushing, K.L., Jackson, C.H., Lockshon,
D., Conover, D., Lanciault, C., Harris, J.R., Simmons, S.J., Rothstein, R. and
Fields, S. (1997). The complete set of predicted genes from Saccharomyces
cerevisiae in a readily usable form.
Genome Res 7: 1169-73.
Hughes, J.D., Estep, P.W., Tavazoie, S. and Church, G.M. (2000).
Computational identification of cis-regulatory elements associated with groups
of functionally related genes in Saccharomyces cerevisiae. J Mol Biol 296(5): 1205-14.
Irizarry, R. A., Hobbs, B., Collin, F., Beazer-Barclay, Y. D.,
Antonellis, K. J., Scherf, U., and Speed, T. P. (2003). Exploration,
normalization, and summaries of high density oligonucleotide array probe level
data. Biostatistics 4: 249-264.
Iyer, V.I., Horak, C.A, Scafe, C.S., Botstein, D., Snyder, M., and
Brown, P.O. (2001). Genomicbinding distribution of the yeast cell-cycle
transcription factors SBF and MBF. Nature 409: 533-538.
Jansen, R., Yu, H., Greenbaum, D., Kluger, Y., Krogan, N.J., Chung, S.,
Emili, A., Snyder, M., Greenblatt, J.F. and Gerstein, M. (2003). A Bayesian networks approach for
predicting protein-protein interactions from genomic data. Science 302: 449-53.
Jensen, S.T. and Liu, J.S. (2004). BioOptimizer: A Bayesian scoring function
approach to motif discovery. Bioinformatics 20: 1557-64.
Ji, H. and Wong, W.H. (2005). TileMap: create chromosomal map of tiling
array hybridizations.
Bioinformatics 21, 3629-3636.
Kent, W.J., Sugnet, C.W., Furey, T.S., Roskin, K.M., Pringle, T.H., Zahler,
A.M. and Haussler D. (2002). The human genome browser at UCSC. Genome Res 12:
996-1006.
Kim, T.H., Barrera, L.O., Zheng, M., Qu, C., Singer, M.A., Richmond,
T.A., Wu, Y., Green, R.D., and Ren, B. (2005). A high-resolution map of active
promoters in the human genome.
Nature 436:876-80.
Kluger, Y., Yu, H., Qian, J., and Gerstein, M. (2003). Relationship
between gene co-expression and probe
localization on microarray slides. BMC Genomics 4: 49.
Lee, T.I., Jenner, R.G., Boyer, L.A., Guenther, M.G., Levine, S.S.,
Kumar, R.M, Chevalier, B., Johnstone, S.E., Cole, M.F., Isono, K., Koseki, H.,
Fuchikami, T., Abe, K., Murray, H.L., Zucker, J.P., Yuan, B., Bell, G.W.,
Herbolsheimer, E., Hannett, N.M., Sun, K., Odom, D.T., Otte, A.P., Volkert,
T.L., Bartel, D.P., Melton, D.A., Gifford D.K., Jaenisch, R., Young, R.A.
(2006). Control of developmental regulators by Polycomb in human embryonic stem
cells. Cell 125: 301-313.
Li, W., Meyer, C.A. and Liu, X.S. (2005). A hidden Markov model for
analyzing ChIP-chip experiments on genome tiling arrays and its application to
p53 binding sequences.
Bioinformatics, 21 Suppl 1: i274-i282.
Liu, X., Brutlag, D.L. and Liu, J.S. (2001). BioProspector: Discovering
conserved DNA motifs in upstream regulatory regions of co-expressed genes. Pac
Symp Biocomput 6: 127-38.
Liu, X.S., Brutlag, D.L. and Liu, J.S. (2002). An algorithm for finding
protein-DNA binding sites with applications to chromatin-immunoprecipitation
microarray experiments. Nat
Biotechnol 20: 835-9.
Liu, Y., Liu, X.S., Wei, L., Altman, R.B. and Batzoglou, S. (2004). Eukaryotic Regulatory Element
Conservation Analysis and Identification Using Comparative Genomics. Genome Res
14: 451–458.
Loots, G., Ovcharenko, I., Pachter, L., Dubchak, I., Rubin, E. (2002).
rVista for comparative sequence-based discovery of functional transcription
factor binding sites. Genome Res 12: 832-839.
Lu, L.J., Xia, Y., Paccanaro, A., Yu, H. and Gerstein, M. (2005). Assessing the limits of genomic data
integration for predicting protein networks. Genome Res 15: 945-53.
Luscombe, N.M., Royce, T.E., Bertone, P., Echols, N., Horak, C.E.,
Chang, J.T., Snyder, M., Gerstein, M. (2003). ExpressYourself: A Modular
Platform for Processing and Visualizing Microarray Data. Nucleic Acids Res
31:3477-3482.
LY Wang, M Snyder and M
Gerstein (2006) BoCaTFBS: a boosted cascade learner to refine the binding sites
suggested by ChIP-chip experiments. Genome Biol, 7, R102.
Martone, R, Euskirchen, G., Bertone, P., Hartman, S., Royce, T.E.,
Luscombe, N.L., John L. Rinn, J.L., Nelson, F.,K., Miller, P., Gerstein, M.,
Weissman, S., Snyder,. M. (2003). Distribution of NF-K B Binding Sites Across
Human Chromosome 22. Proc Natl Acad Sci USA 100: 12247-12252.
Matys, V., Fricke, E., Geffers, R., Gossling, E., Haubrock, M., Hehl,
R., Hornischer, K., Karas, D., Kel, A., Kel-Margoulis, O. (2003). Transfac:
Transcriptional regulation, from patterns to profiles. Nucleic Acids Res 31:
374-378.
Monahan J. (1984) Fast computation of the hodges-lehmann location
estimator. ACM Transactions on Mathematical Software, 10, 270.
Muyrers, J. P., Zhang, Y., and Stewart, A. F. (2000). ET-cloning: Think
recombination first. Genet Eng (N Y) 22: 77-98.
Muyrers, J. P., Zhang, Y., and Stewart, A. F. (2001). Techniques:
Recombinogenic engineering--new options for cloning and manipulating DNA.
Trends Biochem Sci 26: 325-331.
Muyrers, J. P., Zhang, Y., Benes, V., Testa, G., Rientjes, J. M., and
Stewart, A. F. (2004). ET recombination: DNA engineering using homologous
recombination in E. coli. Methods Mol Biol 256: 107-121.
Muyrers, J. P., Zhang, Y., Testa, G., and Stewart, A. F. (1999). Rapid
modification of bacterial artificial chromosomes by ET-recombination. Nucleic
Acids Res 27: 1555-1557.
Nadkarni, P.M., Brandt, C. (1998). Data extraction and ad hoc query of
an entity-attribute-value
Ng, P., Wei, C.L., Sung, W.K., Chiu, K.P., Lipovich, L., Ang, C.C.,
Gupta, S., Shahab, A., Ridwan, A., Wong, C.H., Liu, E.T., Ruan, Y. (2005). Gene
identification signature (GIS) analysis for transcriptome characterization and
genome annotation. Nat. Methods 2: 105-111.
NM Luscombe, MM Babu, H Yu, M Snyder, SA Teichmann, Gerstein, M.
(2004)Genomic analysis of regulatory network dynamics reveals large topological
changes. Nature, 431, 308-12.
Nuwaysir, E.F., Huang, W., Albert, T.J., Singh, J., Nuwaysir, K., Pitas,
A., Richmond, T., Gorski, T., Berg, J.P., Ballin, J., McCormick, M., Norton,
J., Pollock, T., Sumwalt, T., Butcher, L., Porter, D., Molla, M., Hall, C.,
Blattner, F., Sussman, M.R., Wallace, R.L., Cerrina, F., Green, R.D. (2002).
Gene expression analysis using oligonucleotide arrays produced by maskless
photolithography. Genome Res 12: 1749-55.
Pavlidis, P., Furey, T., Liberto, M., and Haussler, D., Grundy, W.
(2001). Promoter region-based classification of genes. Pac Symp Biocomput:
151-163.
Prestridge, D. (1996). Signal Scan 4.0: Additional databases and
sequence formats. Comput Appl
Biosci 12: 157-160.
Ptacek, J., Devgan, G., Michaud, G., Zhu, H., Zhu, X., Fasolo, J., Guo,
H., Jona, G., Breitkreutz, A., Sopko, R., Lee, S., McCartney, R.R., Schmidt,
M.C., Rachidi, N., Stark, M.J.R., Stern, D.F., Tyers, M., de Virgilio, C.,
Andrews, B., Gerstein, M., Schweitzer, B., Predki, P., and Snyder, M.. (2005).
Global Analysis of Protein Phosphorylation in Yeast. Nature 438: 679-84.
Qian, J., Kluger, Y., Yu, H., and Gerstein, M. (2003). Identification
and correction of spurious spatial
correlations in microarray data. Biotechniques 35: 42-4, 46, 48.
Qian, J., Lin, J., Luscombe, N.M., Yu, H. and Gerstein, M. (2003). Prediction
of regulatory networks: genome-wide identification of transcription factor
targets from gene expression data. Bioinformatics 19: 1917-26.
Quandt, K., Frech, K., Karas, H.,
Wingender, E., and Werner, T. (1995). MatInd and MatInspector: new fast and versatile tools for
detection of consensus matches in nucleotide sequence data. Nucleic Acids Res 23: 4878-4884.
Ren, B., Robert, F., Wyrick, J.J., Aparicio, O., Jennings, E.G., Simon,
I., Zeitlinger, J., Schreiber, J., Hannett, N., Kanin, E., Volkert, T.L.,
Wilson, C.J, Bell, S.P. and Young, R.A. (2000). Genome-wide location and
function of DNA binding proteins. Science 290: 2306-9.
Rinn, J.L., Rozowsky, J.S. Laurenz, I.J., Petersen, P.H. Zou, K., Zhong,
W. Gerstein, M., and M. Snyder (2004). Major Molecular Differences Between
Mammalian Sexes are involved in Drug Metabolism and Renal Function. Dev Cell 6:
791-800.
Rinn, J.R., Euskirchen, G., Bertone, P., Martone, R., Luscombe, N.M.,
Hartman, S., Harrison, P.M., Nelson, F.N., Miller, P., Gerstein, M., Weissman,
S., and M. Snyder, (2003). The Transcriptional Activity of Human Chromosome 22,
Genes Dev 17: 529-540.
Ristevski, S. (2005). Making better transgenic models: conditional,
temporal, and spatial approaches. Mol Biotechnol 29: 153-163.
Roulet, E., Busso, S., Camargo, A.A., Simpson, A.J., Mermod, N., and
Bucher, P. (2002). High-throughput
Selex Sage method for quantitative modeling of transcription-factor binding
sites. Nat Biotechnol 20: 831-5
Royce, T. E., Rozowsky, J. S. and Gerstein, M. B. (2007) Assessing the
need for sequence-based normalization in tiling microarray experiments.
Bioinformatics, (in press).
Royce, T.E., Rozowsky, J.S., Bertone, P., Samantac, M., Stolc, V.,
Weissman, S., Snyder, M. and Gerstein, M. (2005). Issues in the Analysis of Oligonucleotide
Tiling Microarrays. Trend Genetics 21: 466-475.
Royce, T.E., Rozowsky, J.S., Luscombe,N.M., Emanuelsson, O., Yu, H.,
Zhu, X., Snyder, M., Gerstein, M. (2006). Extrapolating Traditional DNA
Microarray Statistics to the Tiling and ProteinMicroarray Technologies. Methods
Enzymol: 411. In press.
Rozowsky, J., Newburger, D., Sayward, F., Wu, J., Jordan, G., Korbel1,
J.O., Nagalakshmi, U., Yang, J., Zheng, D., Guigo, R., Gingeras, T., Weissman,
S., Miller, P., Snyder, M., and Gerstein, M. (2006). Analysis and
Classification of Unannotated Transcription within the Encode Regions:
Associating Transcription with Known and Novel Loci. Genome Research. (in
press).
Sandelin, A., Alkema, W., Engstrom, P., Wasserman, W.W., and Lenhard, B.
2004. Jaspar: An open-access
database for eukaryotic transcription factor binding profiles. Nucleic Acids
Res 32: D91-4.
Siepel, A., Bejerano, G., Pedersen, J.S., Hinrichs, A.S., Hou, M., Rosenbloom, K., Clawson, H., Spieth,
J., Hillier, L.W., Richards, S., Weinstock, G.M., Wilson, R.K., Gibbs, R.A.,
Kent, W.J., Miller, W., Haussler,
D. (2005). Evolutionarily conserved elements in vertebrate, insect, worm, and
yeast genomes. Genome Res15: 1034-50.
Smith, A., Greenbaum, D., Douglas, S.M., Long, M. and Gerstein, M.
(2005) Network security and data integrity in academia: an assessment and a
proposal for large-scale archiving. Genome Biol, 6, 119.
Storey, J. D., and Tibshirani, R. (2003). Statistical significance for
genomewide studies. Proc Natl Acad Sci U S A 100: 9440-9445.
Stormo, G. (2000). DNA binding sites: Representation and discovery.
Bioinformatics 16: 16-23.
Trinklein, N.D., Karaöz, U., Wu, J., Halees, A., Aldred, S.F., Collins,
P.J., Zheng, D., Zhang, Z., Gerstein, M., Snyder, M., Myers, R.M. and Weng, Z.
(2006). Genome Research. In press.
Troyanskaya, O.G., Garber, M.E., Brown, P.O., Botstein, D. and Altman,
R.B. (2002). Nonparametric methods for identifying differentially expressed
genes in microarray data Bioinformatics 18: 1454-1461.
Tusher, V. G., Tibshirani, R., and Chu, G. (2001). Significance analysis
of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U
S A 98: 5116-5121.
Urban, A.E., Korbel, J., Selzer, R., Popescu, G.V., Richmond, T.,
Cubells, J.F., Green, R., Emanuel B.S., Gerstein, M., Weissman, S.M., and
Snyder, M. (2006) High Resolution Mapping of DNA Copy Alterations Using High
Density Tiling Oligonucleotide Arrays. Proc Natl Acad Sci 103: 4534-4539.
van Steensel, B., Delrow, J. and Henikoff, S. (2001). Chromatin
profiling using targeted DNA adenine methyltransferase. Nat Genet 27: 304-8.
Vlieghe, D., Sandelin, A., De Bleser, P.J., Vleminckx, K., Wasserman,
W.W., van Roy, F. and Lenhard, B. (2006). A new generation of Jaspar, the
open-access repository for transcription factor binding site profiles. Nucleic
Acids Res. 34: D95-7.
Washietl, S., Pedersen, J. S., Korbel, J. O., Gruber, A. R.,
Hackermuller, J., Hertel, J., Lindemeyer, M., Reiche, K., Stocsits, C., Tanzer,
A., Ucla, C., Wyss, C., Antonarakis, S. E., Denoeud, F., Lagarde, J., Drenkow,
J., Kapranov, P., Gingeras, T. R., Snyder, M., Gerstein, M. B., Reymond, A.,
Hofacker, I. L., Stadler, P. F. (2007) Structured RNAs in the ENCODE Selected
Regions of the Human Genome. Genome Research, (in press)
Wasserman, W., and Sandelin, A. (2004). Applied Bioinformatics for the
identification of regulatory elements. Nature 5: 278-287.
Wei, Z. and Jensen, S.T. (2006 ).
GAME: detecting cis-regulatory elements using a genetic algorithm.
Bioinformatics 22: 1577-1584.
Workman, C.T., Mak, H.C., McCuine, S., Tagne, J.B., Agarwal, M., Ozier,
O., Begley, T.J., Samson, L.D. and Ideker, T. (2006). A systems approach to mapping DNA damage response pathways.
Science 312: 1054-9.
Yip, K. Y., Yu, H., Kim, P. M., Schultz, M. and Gerstein, M. (2006) The
tYNA platform for comparative interactomics: a web tool for managing, comparing
and mining multiple networks. Bioinformatics, 22, 2968-70.
Yu, H. and Gerstein, M. (2006) Genomic analysis of the hierarchical
structure of regulatory networks. Proc Natl Acad Sci U S A, 103, 14724-31.
Yu, H., Luscombe, N. M., Lu, H. X., Zhu, X., Xia, Y., Han, J. D.,
Bertin, N., Chung, S., Vidal, M. and
Gerstein, M. (2004) Annotation transfer between genomes: protein-protein
interologs and protein-DNA regulogs. Genome Res, 14, 1107-18.
Yu, H., Nguyen, K., Royce, T., Qian, J., Nelson, K., Snyder, M. and
Gerstein, M. (2007) Positional artifacts in microarrays: experimental
verification and construction of COP, an automated detection tool. Nucleic
Acids Res, 35, e8.
Yu, H., Zhu, X., Greenbaum, D., Karro, J. and Gerstein, M. (2004)
TopNet: a tool for comparing biological sub-networks, correlating protein
properties with topological statistics. Nucleic Acids Res, 32, 328-37.
Zhang, Z., Harrison, P.M., Liu, Y., and Gerstein, M. (2003). Millions of
years of evolution preserved: a comprehensive catalog of the processed
pseudogenes in the human genome. Genome Res 13: 2541-58.
Zhang, Z., Paccanaro,A., Fu, Y.,
Weissman, S., Weng, Z., Chang, J., Snyder, M., Gerstein, M. (2006). Development of approaches for global analysis of regulatory
elements global analysis of the genomic distribution and correlation of
regulatory elements in the encode regions. Genome Research. (in press)
Zhang, Z., Pang, A. W. and Gerstein, M. (2007) Comparative analysis of
genome tiling array data reveals many novel primate-specific functional RNAs in
human. BMC Evol Bio, 7, Suppl 1, S14.
Zheng, D. and Gerstein, M. B. (2006) A computational approach for
identifying pseudogenes in the ENCODE regions. Genome Biol 7 Suppl 1, S13,
1-10.
Zheng, D. and Gerstein, M. B. (2007).The ambiguous boundary between
genes and pseudogenes: the dead rise up, or do they? Trends in Genetics. In
press
Zheng, D., Adam Frankish, A., Baertsch, R., Kapranov, P., Reymond, A.,
Choo, S.W., Lu, Y., Denoeud, F.,
Antonarakis, S.E., Snyder, M., Ruan, Y., Wei, C., Gingeras, T.R., Guigo, R.,
Harrow, J., and Gerstein, M. (2007). Pseudogenes in the Encode Regions:
Consensus Annotation, Analysis of Transcription and Evolution. Genome Research.
in press.
Zheng, D., Zhang, Z., Harrison, P.M., Karro, J., Carriero, N., and
Gerstein, M. (2005). Integrated pseudogene annotation for human chromosome 22:
evidence for transcription. J Mol Biol 349: 27-45.