 A
second option for a general lack of correlation between mRNA and
protein abundance
may be that proteins have very different half-lives as the result of
varied
protein synthesis and degradation. Protein turnover can vary
significantly
depending on a number of different conditions (Glickman, 2002); the
cell can
control the rates of degradation or synthesis for a given protein, and
there
is significant heterogeneity even within proteins that have similar
functions
(Pratt, 2002). Recent efforts have been made to computationally
measure these
rates (Lian, 2002).
A
second option for a general lack of correlation between mRNA and
protein abundance
may be that proteins have very different half-lives as the result of
varied
protein synthesis and degradation. Protein turnover can vary
significantly
depending on a number of different conditions (Glickman, 2002); the
cell can
control the rates of degradation or synthesis for a given protein, and
there
is significant heterogeneity even within proteins that have similar
functions
(Pratt, 2002). Recent efforts have been made to computationally
measure these
rates (Lian, 2002).
Simplistically, it can be presumed that the change in a protein's
concentration
over time will be equal to the rate of translation minus the rate of
degradation.
By analogy to concepts in chemical kinetics, we can approximate this
equation:
dP(i,t)/dt = SE(i,t) - DP(i,t), where P is protein abundance i at time
t,
E is the mRNA expression level of protein P, S is a general rate of
protein
synthesis per mRNA, and D is a general rate of protein degradation per
protein
(Gerner, 2002). Additionally there are some experimental methods that
can
also be used to measure turnover and the translational control of
protein
levels (e.g., Serikawa, 2003). Given the degenerate nature of the
genetic
code, there are many synonymous codons (codons that translate into the
same
amino acid). As the cell is biased in its usage of synonymous codons -
that
is, the usage of a subset of codons results in a higher level of mRNA
expression,
possibly as a result of differing cellular tRNA levels - the CAI can
be used
to predict the expression of a gene (Sharp, 1987). We recently
calculated
new parameters for this model, with some improvement in predictive
strength
(Jansen, 2003). It is thought that the CAI will correlate differently
with
mRNA levels than with protein abundance levels due, in part, to
protein turnover
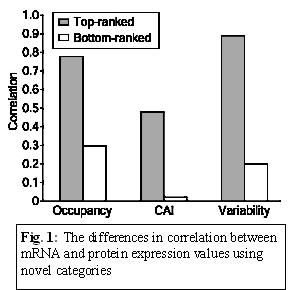
rates (Coghlan, 2000). Ranking the ORFs in terms of their CAI value,
we found
that although those ORFs that ranked the highest in terms of CAI did
not show
a very strong correlation between mRNA and protein levels, they
nevertheless
showed a significantly higher correlation than ORFs that were ranked
as having
the lower CAI values (r = 0.48 versus 0.02). The low correlations
reflect
the fact that the CAI will correlate differently for protein and mRNA
values
because of the additional cellular controls on protein translation,
namely
the effect of protein turnover rates. Nevertheless, the sizable
difference
in correlations between the two groups of ORFs with high- and
low-ranking
CAI values (Figure 1) shows there is some relationship between mRNA
and protein
values, possibly indicating that highly expressed genes tend to result
in
a more correlated level of protein abundance than lower expressed
ones.
Although proteomics is still in its infancy, given the pace of
technological
advancement in protein quantification, mRNA expression analysis and
noise
reduction, more comprehensive correlation studies will soon be
feasible. This
will allow for more robust analyses of the relationship between mRNA
expression
and protein abundance values. Indeed, we are in the process of
continuing
this line of research by careful examination and correlation of the
mRNA expression
data already obtained by R. Duman and other Yale neurobiologists in
the proposed
Center of Excellence in Neuroproteomics. One obvious goal of these
studies
will be to be able to reach the point where we can more accurately
extrapolate
mRNA to protein expression data and thereby, for instance, guide the
selection
of antibodies to be spotted onto microarrays and those proteins that
will
be targeted by directed MS/MS-based technologies to permit the
independent
measurement of selected proteins of high potential interest.
Literature Cited
Anderson L, Seilhamer J (1997) A comparison of selected mRNA and
protein abundances
in human liver. Electrophoresis 18: 533-537.
Arava Y, Wang Y, Storey JD, Liu CL, Brown PO, Herschlag D (2003)
Genome-wide
analysis of mRNA translation profiles in Saccharomyces cerevisiae. Proc
Natl
Acad Sci USA 100: 3889-3894.
Arthur, JM (2003) Proteomics : Curr Opin Nephrol Hypertens. 12(4):
423-30.
Bader, G.D. and Hogue, C.W. (2000). BINDA data specification for storing
and
describing biomolecular interactions, molecular complexes and pathways.
Bioinformatics
16: 465-477.
Chen G, Gharib TG, Huang CC, Taylor JM, Misek DE, Kardia SL, Giordano
TJ, Iannettoni
MD, Orringer MB, Hanash SM, et al. (2002) Discordant protein and mRNA
expression
in lung adenocarcinomas. Mol Cell Proteomics 1:304-313.
Chothia, C. and Gerstein, M. (1997) Protein evolution. How far can
sequences
diverge? Nature 385 : 579, 581.
Coghlan A, Wolfe KH (2000) Relationship of codon bias to mRNA
concentration
and protein length in Saccharomyces cerevisiae.Yeast 16:1131-1145.
Day, D. A. and M. F. Tuite (1998). Post-transcriptional gene regulatory
mechanisms
in eukaryotes:an overview. J Endocrinol 157(3): 361-71.
Drawid A, Gerstein M. (2000) A Bayesian system integrating expression
data with
sequence patterns for localizing proteins: comprehensive application to
the
yeast genome. J Mol Biol. 301(4):1059-75.
Futcher B, Latter GI, Monardo P, McLaughlin CS, Garrels JI (1999) A
sampling
of the yeast proteome. Mol Cell Biol 19:7357-7368.
Gerner C, Vejda S, Gelbmann D, Bayer E, Gotzmann J, Schulte-Hermann R,
Mikulits
W (2002) Concomitant determination of absolute values of cellular
protein amounts,
synthesis rates, and turnover rates by quantitative proteome profiling.
Mol
Cell Proteomics 1:528-537.
Gerstein, M. (1998) Measurement of the effectiveness of transitive
sequence
comparison, through a third 'intermediate' sequence. Bioinformatics 14 :
707-14.
Gerstein, M. and Jansen, R. (2000). The current excitement in
bioinformatics-analysis
of whole- genome expression data: how does it relate to protein
structure and
function?
Curr Opin Struct Biol 10 : 574-84.
Glickman MH, Ciechanover A (2002) The ubiquitin-proteasome proteolytic
pathway:
destruction for the sake of construction. Physiol Rev 82:373-428.
Greenbaum, D.,Colangelo, C., Williams, K., and Gerstein, M. (2003)
Comparing
protein abundance and mRNA expression levels on a genomic scale. Genome
Biology,
4, 117.1-117.8.
Greenbaum D, Jansen R, Gerstein M (2002) Analysis of mRNA expression and
protein
abundance data: an approach for the comparison of the enrichment of
features
in the cellular population of proteins and transcripts. Bioinformatics
18:585-596.
Greenbaum D, Luscombe NM, Jansen R, Qian J, Gerstein M (2001)
Interrelating
different types of genomic data, from proteome to secretome: 'oming in
on function.
Genome Res 11:1463-1468.
Gygi, SP, Corthals, GL, Zhang, Y, Rochon, Y & Aebersold, R (2000)
Evaluation
of two-dimensional gel electrophoresis-based proteome analysis
technology. Proc
Natl Acad Sci USA, 97, 9390-5.
Gygi, SP, Rochon, Y, Franza, BR & Aebersold, R (1999). Correlation
between
protein and mRNA abundance in yeast. Mol Cell Biol 19,1720-30.
Hegyi H, Gerstein M. (2001) Annotation transfer for genomics: measuring
functional
divergence in multi-domain proteins. Genome Res.11(10):1632-40.
Holstege, F. C., E. G. Jennings, et al. (1998). Dissecting the
regulatory circuitry
of a eukaryotic genome. Cell 95(5): 717-728.
Ito, T., Chiba, T., Ozawa, R., Yoshida, M., Hattori, M., and Sakaki,
Y.(2001).
A comprehensive two-hybrid analysis to explore the yeast protein
interactome.
Proc. Natl. Acad. Sci. 98: 4569-4574.
Jacobs Anderson JS, Parker R. Computational identification of cis-acting
elements
affecting post transcriptional control of gene expression in
Saccharomyces cerevisiae.
Nucleic Acids Res. 2000 Apr 1;28(7):1604-17.
Jackson, R. J. and M. Wickens (1997). Translational controls impinging
on the
5'-untranslated region and initiation factor proteins. Curr Opin Genet
Dev 7(2):
233-41.
Jansen R, Yu H, Greenbaum D, Kluger Y, Krogan NJ, Chung S, Emili A,
Snyder M,
Greenblatt JF, Gerstein M. (2003) A bayesian networks approach for
predicting
protein-protein interactions from genomic data. Science.
302(5644):449-53.
Jansen R, Bussemaker HJ, Gerstein M (2003) Revisiting the codon
adaptation index
from a whole-genome perspective: analyzing the relationship between gene
expression
and codon occurrence in yeast using a variety of models. Nucleic Acids
Res 31:2242-2251.
Jansen R, Greenbaum D, Gerstein M (2002) Relating whole-genome
expression data
with protein-protein interactions. Genome Res 12:37-46.
Jansen R, Lan N, Qian J, Gerstein M. (2002) Integration of genomic
datasets
to predict protein complexes in yeast J Struct Funct Genomics.
2002b;2(2):71-81.
Jelinsky, S. A. and L. D. Samson (1999). Global response of
Saccharomyces cerevisiae
to an alkylating agent. Proc Natl Acad Sci U S A 96(4): 1486-91.
Lian Z, Kluger Y, Greenbaum DS, Tuck D, Gerstein M, Berliner N, Weissman
SM,
Newburger PE (2002) Genomic and proteomic analysis of the myeloid
differentiation
program: global analysis of gene expression during induced
differentiation in
the MPRO cell line. Blood 100:3209-3220.
Lindahl, L. and A. Hinnebusch (1992). Diversity of mechanisms in the
regulation
of translation in prokaryotes and lower eukaryotes. Curr Opin Genet Dev
2(5):
720-6.
McCarthy, J. E. (1998). Posttranscriptional control of gene expression
in yeast.
Microbiol Mol Biol Rev 62(4): 1492-553.
Mewes HW, Frishman D, Guldener U, Mannhaupt G, Mayer K, Mokrejs M,
Morgenstern
B, Munsterkotter M, Rudd S, Weil B. (2002) MIPS: a database for genomes
and
protein sequences. Nucleic Acids Res. 30(1):31-4.
Morris, D. R. and A. P. Geballe (2000). Upstream open reading frames as
regulators
of mRNA translation. Mol Cell Biol 20(23): 8635-42.
Naylor, G. J. and Gerstein, M. (2000) Measuring shifts in function and
evolutionary
opportunity using variability profiles: A case study of the globins. J
Mol Evol
51: 223-33.
Orntoft TF, Thykjaer T, Waldman FM, Wolf H, Celis JE (2002) Genome-wide
study
of gene copy numbers, transcripts, and protein levels in pairs of
non-invasive
and invasive human transitional cell carcinomas. Mol Cell Proteomics 1:
37-45.
Peng J, Elias JE, Thoreen CC, Licklider LJ, Gygi SP (2003) Evaluation of
multidimensional
chromatography coupled with tandem mass spectrometry (LC/LC-MS/MS) for
large-scale
protein analysis: the yeast proteome. J Proteome Res 2: 43-50.
Pratt JM, Petty J, Riba-Garcia I, Robertson DH, Gaskell SJ, Oliver SG,
Beynon
RJ (2002) Dynamics of protein turnover, a missing dimension in
proteomics. Mol
Cell Proteomics 1:579-591.
Qian J, Lin J, Luscombe NM, Yu H, Gerstein M. (2003) Prediction of
regulatory
networks: genome-wide identification of transcription factor targets
from gene
expression data. Bioinformatics.19(15):1917-26.
Roth, F. P., J. D. Hughes, et al. (1998). Finding DNA regulatory motifs
within
unaligned noncoding sequences clustered by whole-genome mRNA
quantitation. Nat
Biotechnology 16(10): 939-45.
Serikawa KA, Xu XL, MacKay VL, Law GL, Zong Q, Zhao LP, Bumgarner R,
Morris
DR (2003) The transcriptome and its translation during recovery from
cell cycle
arrest in Saccharomyces cerevisiae. Mol Cell Proteomics 2: 191-204.
Sharp PM, Li WH (1987) The codon adaptation index --- a measure of
directional
synonymous codon usage bias, and its potential applications. Nucleic
Acids Res
15:1281- 1295.
Uetz, P., Giot, L., Cagney, G., Mansfield, T.A., Judson, R.S., Knight,
J.R.,
Lockshon, D., Narayan, V., Srinivasan, M., Pochart, P. (2000). A
comprehensive
analysis of protein-protein interactions in Saccharomyces cerevisiae.
Nature
403: 623-627.
Velculescu VE, Zhang L, Zhou W, Vogelstein J, Basrai MA, Bassett DE Jr,
Hieter
P, Vogelstein B, Kinzler KW (1997). Characterization of the yeast
transcriptome.
Cell 88(2):243-51.
Vilela, C., B. Linz, et al. (1998). The yeast transcription factor genes
YAP1
and YAP2 are subject to differential control at the levels of both
translation
and mRNA stability. Nucleic AcidsRes 26(5): 1150-9.
Vilela, C., C. V. Ramirez, et al. (1999). Post-termination ribosome
interactions
with the 5'UTR modulate yeast mRNA stability. Embo J 18(11):
3139-52.
Washburn MP, Wolters D, Yates 3rd JR (2001) Large-scale analysis of the
yeast
proteome by multidimensional protein identification technology. Nat
Biotechnol
19:242-247.
Washburn MP, Koller A, Oshiro G, Ulaszek RR, Plouffe D, Deciu C,
Winzeler E,
Yates 3rd JR (2003) Protein pathway and complex clustering of correlated
mRNA
and protein expression analyses in Saccharomyces cerevisiae. Proc Natl
Acad
Sci USA 100:3107-3112.
Wilson CA, Kreychman J, Gerstein M. (2000) Assessing annotation transfer
for
genomics: quantifying the relations between protein sequence, structure
and
function through traditional and probabilistic scores. J Mol Biol.
297(1):233-49.
Xenarios, I., Rice, D.W., Salwinski, L., Baron, M.K., Marcotte, E.M.,
and Eisenberg,
D. (2000). DIP: The database of interacting proteins. Nucleic Acids Res.
28:
289-291.