What is bioinformatics? An
introduction and overview
Nicholas M Luscombe, Dov
Greenbaum & Mark Gerstein*
Department of Molecular
Biophysics and Biochemistry
Yale University, 266 Whitney
Avenue
PO Box 208114, New Haven CT
06520-8114, USA
* - corresponding author
For IMIA
2001 Yearbook
Click here for PDF version
1. Abstract
A flood of data means that many of the challenges in biology are now challenges in computing. Bioinformatics, the application of computational techniques to analyse the information associated with biomolecules on a large-scale, has now firmly established itself as a discipline in molecular biology, and encompasses a wide range of subject areas from structural biology, genomics to gene expression studies.
In this review we provide an
introduction and overview of the current state of the field. We discuss the
main principles that underpin bioinformatics analyses, look at the types of
biological information and databases that are commonly used, and finally
examine some of the studies that are being conducted, particularly with
reference to transcription regulatory systems.
2. Introduction
Biological data are flooding in at an unprecedented rate (1). For example as of August 2000, the GenBank repository of nucleic acid sequences contained 8,214,000 entries (2) and the SWISS-PROT database of protein sequences contained 88,166 (3). On average, the amount of information stored in these databases is doubling every 15 months (2). In addition, since the publication of the H. influenzae genome (4), complete sequences for over 40 organisms have been released, ranging from 450 genes to over 100,000. Add to this the data from the myriad of related projects that study gene expression, determine the protein structures encoded by the genes, and detail how these products interact with one another, and we can begin to imagine the enormous quantity and variety of information that is being produced.
2.1 Bioinformatics – a definition
(Molecular)
bio – informatics: bioinformatics is conceptualising biology in
terms of molecules (in the sense of physical chemistry) and applying “informatics
techniques” (derived from disciplines such as applied maths,
computer science and statistics) to understand and organise
the information associated with these molecules, on a large
scale. In short, bioinformatics is a management information system
for molecular biology and has many practical applications.
As a result of this surge in data, many of the challenges in biology have actually become challenges in computing. Such an approach is ideal because of the ease with which computers can handle large quantities of data and probe the complex dynamics observed in nature. Bioinformatics, the subject of the current review, is often defined as the application of computational techniques to understand and organise the information associated with biological macromolecules. This shotgun marriage between the two subjects is largely attributed to the fact that biology itself is an information technology; an organism’s physiology and behaviour are largely dictated by its genes, which at the basic level can be viewed as digital repositories of information. At the same time, there have been major advances in the technologies that supply the raw data; according to Anthony Kerlavage of Celera, an experimental laboratory can easily produce over 100 gigabytes of data a day (5). This incredible processing power has been matched by developments in computer technology; the most important areas of improvements have been in the CPU, disk storage and Internet, allowing faster computations, better data storage and revolutionalised the methods for accessing and exchanging of data.
2.2 Aims of bioinformatics
The aims of bioinformatics are three-fold. First, at its simplest bioinformatics organises data in a way that allows researchers to access existing information and to submit new entries as they are produced, eg the Protein Data Bank for 3D macromolecular structures (6, 7). While data-curation is an essential task, the information stored in these databases is essentially useless until analysed. Thus the purpose of bioinformatics extends far beyond mere volume control. The second aim is to develop tools and resources that aid in the analysis of data. For example, having sequenced a particular protein, it is of interest to compare it with previously characterised sequences. This requires more than just a straightforward database search. As such, programs such as FASTA (8) and PSI-BLAST (9) must consider what constitutes a biologically significant resemblance. Development of such resources requires extensive knowledge of computational theory, as well as a thorough understanding of biology. The third aim is to use these tools to analyse the data and interpret the results in a biologically meaningful manner. Traditionally, biological studies examined individual systems in detail, and frequently compared them with a few that are related. In bioinformatics, we can also conduct global analyses of all the available data with the aim of uncovering common principles that apply across many systems and highlight features that are unique to some.
In this review, we provide an introduction to bioinformatics. We focus on the first and third aims just described, with particular reference to the keywords underlined in the definition: information, informatics, organisation, understanding, large-scale and practical applications. Specifically, we discuss the range of data that are currently being examined, the databases into which they are organised, the types of analyses that are being conducted using transcription regulatory systems as an example, and finally discuss some of the major practical applications of bioinformatics.
3. "...the INFORMATION associated with these molecules..."
Table 1 lists the types of data that are analysed in bioinformatics and the range of topics that we consider to fall within the field. Here we take a broad view and include subjects that may not normally be listed. We also give approximate values describing the sizes of data being discussed.
Table
1. Sources of
data used in bioinformatics, the quantity of each type of data that is
currently (August 2000) available, and bioinformatics subject areas that
utilise this data.
We
start with an overview of the sources of information: these may be divided into
raw DNA sequences, protein sequences, macromolecular structures, genome
sequences, and other whole genome data. Raw DNA sequences are strings of the
four base-letters comprising genes, each typically 1,000 bases long. The
GenBank repository of nucleic acid sequences currently holds a total of 9.5 billion bases in 8.2 million
entries (all database figures as of August 2000). At the next level are protein
sequences comprising strings of 20 amino acid-letters. At present there are
about 300,000 known protein sequences, with a typical bacterial protein
containing approximately 300 amino acids. Macromolecular structural data
represents a more complex form of information. There are currently 13,000
entries in the Protein Data Bank, PDB, most of which are protein structures. A
typical PDB file for a medium-sized protein contains the xyz coordinates of
approximately 2,000 atoms.
Scientific euphoria has recently
centred on whole genome sequencing. As with the raw DNA sequences, genomes
consist of strings of base-letters, ranging from 1.6 million bases in Haemophilus
influenzae to 3 billion in humans. An important aspect of complete genomes
is the distinction between coding regions and non-coding regions –‘junk’
repetitive sequences making up the bulk of base sequences especially in
eukaryotes. We can now measure expression levels of almost every gene in a
given cell on a whole-genome level although public availability of such data is
still limited. Expression level measurements are made under different
environmental conditions, different stages of the cell cycle and different cell
types in multi-cellular organisms. Currently the largest dataset for yeast has
made approximately 20 time-point measurements for 6,000 genes (10). Other genomic-scale data include biochemical
information on metabolic pathways, regulatory networks, protein-protein
interaction data from two-hybrid experiments, and systematic knockouts of
individual genes to test the viability of an organism.
What is apparent from this list is the diversity in the size and complexity of different datasets. There are invariably more sequence-based data than structural data because of the relative ease with which they can be produced. This is partly related to the greater complexity and information-content of individual structures compared to individual sequences. While more biological information can be derived from a single structure than a protein sequence, the problem is overcome in the latter by analysing larger quantities of data.
4. "...ORGANISE the information on a LARGE SCALE
..."
4.1 Redundancy and multiplicity of data
A concept that underpins most research methods in bioinformatics is that much of this data can be grouped together based on biologically meaningful similarities. For example, sequence segments are often repeated at different positions of genomic DNA (11). Genes can be clustered into those with particular functions (eg enzymatic actions) or according to the metabolic pathway to which they belong (12), although here, single genes may actually possess several functions (13). Going further, distinct proteins frequently have comparable sequences – organisms often have multiple copies of a particular gene through duplication and different species have equivalent or similar proteins that were inherited when they diverged from each other in evolution. At a structural level, we predict there to be a finite number of different tertiary structures – estimates range between 1,000 and 10,000 folds (14, 15) – and proteins adopt equivalent structures even when they differ greatly in sequence (16). As a result, although the number of structures in the PDB has increased exponentially, the rate of discovery of novel folds has actually decreased.
There are common terms to describe the relationship between pairs of proteins or the genes from which they are derived: analogous proteins have related folds, but unrelated sequences, while homologous proteins are both sequentially and structurally similar. The two categories can sometimes be difficult to distinguish especially if the relationship between the two proteins is remote (17, 18). Among homologues, it is useful to distinguish between orthologues, proteins in different species that have evolved from a common ancestral gene, and paralogues, proteins that are related by gene duplication within a genome (19). Normally, orthologues retain the same function while paralogues evolve distinct, but related functions (20).
An important concept that arises from these observations is that of a finite “parts list” for different organisms (21, 22): an inventory of proteins contained within an organism, arranged according to different properties such as gene sequence, protein fold or function. Taking protein folds as an example, we mentioned that with a few exceptions, the tertiary structures of proteins adopt one of a limited repertoire of folds. As the number of different fold families is considerably smaller than the number of gene families, categorising the proteins by fold provides a substantial simplification of the contents of a genome. Similar simplifications can be provided by other attributes such as protein function. As such, we expect this notion of a finite parts list to become increasingly common in the future genomic analyses.
Clearly, an essential aspect of managing this large volume of data lies in developing methods for assessing similarities between different biomolecules and identifying those that are related. Below, we discuss the major databases that provide access to the primary sources of information, and also introduce some secondary databases that systematically group the data (Table 2). These classifications ease comparisons between genomes and their products, allowing the identification of common themes between those that are related and highlighting features that are unique to some.
Table
2. List of
URLs for the databases that are cited in the review.
4.2 Protein sequence databases
Protein sequence databases are categorised as primary, composite or secondary. Primary databases contain over 300,000 protein sequences and function as a repository for the raw data. Some more common repositories, such as SWISS-PROT (3) and PIR-International (23), annotate the sequences as well as describe the proteins’ functions, its domain structure and post-translational modifications. Composite databases such as OWL (24) and the NRDB (25) compile and filter sequence data from different primary databases to produce combined non-redundant sets that are more complete than the individual databases and also include protein sequence data from the translated coding regions in DNA sequence databases (see below). Secondary databases contain information derived from protein sequences and help the user determine whether a new sequence belongs to a known protein family. One of the most popular is PROSITE (26), a database of short sequence patterns and profiles that characterise biologically significant sites in proteins. PRINTS (27) expands on this concept and provides a compendium of protein fingerprints – groups of conserved motifs that characterise a protein family. Motifs are usually separated along a protein sequence, but may be contiguous in 3D-space when the protein is folded. By using multiple motifs, fingerprints can encode protein folds and functionalities more flexibly than PROSITE. Finally, Pfam (28) contains a large collection of multiple sequence alignments and profile Hidden Markov Models covering many common protein domains. Pfam-A comprises accurate manually compiled alignments while Pfam-B is an automated clustering of the whole SWISS-PROT database. These different secondary databases have recently been incorporated into a single resource named InterPro (29).
4.3 Structural databases
Next we look at databases of
macromolecular structures. The Protein Data Bank, PDB (6, 7), provides a primary archive of all 3D structures for
macromolecules such as proteins, RNA, DNA and various complexes. Most of the
~13,000 structures (August 2000) are solved by x-ray crystallography and NMR,
but some theoretical models are also included. As the information provided in
individual PDB entries can be difficult to extract, PDBsum (30) provides a separate Web page for every structure in
the PDB displaying detailed structural analyses, schematic diagrams and data on
interactions between different molecules in a given entry. Three major
databases classify proteins by structure in order to identify structural and
evolutionary relationships: CATH (31), SCOP (32), and FSSP databases (33). All comprise hierarchical structural taxonomy where
groups of proteins increase in similarity at lower levels of the classification
tree. In addition, numerous databases focus on particular types of macromolecules.
These include the Nucleic Acids Database, NDB (34), for structures related to nucleic acids, the HIV
protease database (35) for HIV-1, HIV-2 and SIV protease structures and
their complexes, and ReLiBase (36) for receptor-ligand complexes.
4.4 Nucleotide and Genome sequences
As described previously, the biggest excitement currently lies with the availability of complete genome sequences for different organisms. The GenBank (2), EMBL (37) and DDBJ (38) databases contain DNA sequences for individual genes that encode protein and RNA products. Much like the composite protein sequence database, the Entrez nucleotide database (39) compiles sequence data from these primary databases.
As whole-genome sequencing is often conducted through international collaborations, individual genomes are published at different sites. The Entrez genome database (40) brings together all complete and partial genomes in a single location and currently represents over 1,000 organisms (August 2000). In addition to providing the raw nucleotide sequence, information is presented at several levels of detail including: a list of completed genomes, all chromosomes in an organism, detailed views of single chromosomes marking coding and non-coding regions, and single genes. At each level there are graphical presentations, pre-computed analyses and links to other sections of Entrez. For example, annotations for single genes include the translated protein sequence, sequence alignments with similar genes in other genomes and summaries of the experimentally characterised or predicted function. GeneCensus (41) also provides an entry point for genome analysis with an interactive whole-genome comparison from an evolutionary perspective. The database allows building of phylogenetic trees based on different criteria such as ribosomal RNA or protein fold occurrence. The site also enables multiple genome comparisons, analysis of single genomes and retrieval of information for individual genes. The COGs database (20) classifies proteins encoded in 21 completed genomes on the basis of sequence similarity. Members of the same Cluster of Orthologous Group, COG, are expected to have the same 3D domain architecture and often, similar functions. The most straightforward application of the database is to predict the function of uncharacterised proteins through their homology to characterised proteins, and also to identify phylogenetic patterns of protein occurrence – for example, whether a given COG is represented across most or all organisms or in just a few closely related species.
4.5 Gene expression data
The most recent sources of genomic-scale data have been from expression experiments, which quantify the expression levels of individual genes. These experiments measure the amount of mRNA or protein products that are produced by the cell. For the former, there are three main technologies: the cDNA microarray (42-44), Affymatrix GeneChip (45) and SAGE methods (46). The first method measures relative levels of mRNA abundance between different samples, while the last two measure absolute levels. Most of the effort in gene expression analysis has concentrated on the yeast and human genomes and as yet, there is no central repository for this data. For yeast, the Young (10), Church (47) and Samson datasets (48) use the GeneChip method, while the Stanford cell cycle (49), diauxic shift (50) and deletion mutant datasets (51) use the microarray. Most measure mRNA levels throughout the whole yeast cell cycle, although some focus on a particular stage in the cycle. For humans, the main application has been to understand expression in tumour and cancer cells. The Molecular Portraits of Breast Tumours (52), Lymphoma and Leukaemia Molecular Profiling (53) projects provide data from microarray experiments on human cancer cells.
The technologies for measuring protein abundance are currently limited to 2D gel electrophoresis followed by mass spectrometry (54). As gels can only routinely resolve about 1,000 proteins (55), only the most abundant can be visualised. At present, data from these experiments are only available from the literature (56, 57).
4.6 Data integration
The most profitable research in bioinformatics often results from integrating multiple sources of data (58). For instance, the 3D coordinates of a protein are more useful if combined with data about the protein’s function, occurrence in different genomes, and interactions with other molecules. In this way, individual pieces of information are put in context with respect to other data. Unfortunately, it is not always straightforward to access and cross-reference these sources of information because of differences in nomenclature and file formats.
At a basic level, this problem is frequently addressed by providing external links to other databases, for example in PDBsum, web-pages for individual structures direct the user towards corresponding entries in the PDB, NDB, CATH, SCOP and SWISS-PROT. At a more advanced level, there have been efforts to integrate access across several data sources. One is the Sequence Retrieval System, SRS (59), which allows any flat-file databases to be indexed to each other; this allows the user to retrieve, link and access entries from nucleic acid, protein sequence, protein motif, protein structure and bibliographic databases. Another is the Entrez facility (39), which provides similar gateways to DNA and protein sequences, genome mapping data, 3D macromolecular structures and the PubMed bibliographic database (60). A search for a particular gene in either database will allow smooth transitions to the genome it comes from, the protein sequence it encodes, its structure, bibliographic reference and equivalent entries for all related genes.
5. "...UNDERSTAND and organise the information..."
Having examined the data, we can discuss the types of analyses that are conducted. As shown in Table 1, the broad subject areas in bioinformatics can be separated according to the sources of information that are used in the studies. For raw DNA sequences, investigations involve separating coding and non-coding regions, and identification of introns, exons and promoter regions for annotating genomic DNA (61) (62). For protein sequences, analyses include developing algorithms for sequence comparisons (63), methods for producing multiple sequence alignments (64), and searching for functional domains from conserved sequence motifs in such alignments. Investigations of structural data include prediction of secondary and tertiary protein structures, producing methods for 3D structural alignments (65, 66), examining protein geometries using distance and angular measurements, calculations of surface and volume shapes and analysis of protein interactions with other subunits, DNA, RNA and smaller molecules. These studies have lead to molecular simulation topics in which structural data are used to calculate the energetics involved in stabilising macromolecular structures, simulating movements within macromolecules, and computing the energies involved in molecular docking. The increasing availability of annotated genomic sequences has resulted in the introduction of computational genomics and proteomics – large-scale analyses of complete genomes and the proteins that they encode. Research includes characterisation of protein content and metabolic pathways between different genomes, identification of interacting proteins, assignment and prediction of gene products, and large-scale analyses of gene expression levels. Some of these research topics will be demonstrated in our example analysis of transcription regulatory systems.
Other subject areas we have included in Table 1 are development of digital libraries for automated bibliographical searches, knowledge bases of biological information from the literature, DNA analysis methods in forensics, prediction of nucleic acid structures, metabolic pathway simulations, and linkage analysis – linking specific genes to different disease traits.
In addition to finding relationships between different proteins, much of bioinformatics involves the analysis of one type of data to infer and understand the observations for another type of data. An example is the use of sequence and structural data to predict the secondary and tertiary structures of new protein sequences (67). These methods, especially the former, are often based on statistical rules derived from structures, such as the propensity for certain amino acid sequences to produce different secondary structural elements. Another example is the use of structural data to understand a protein’s function; here studies have investigated the relationship different protein folds and their functions (68, 69) and analysed similarities between different binding sites in the absence of homology (70). Combined with similarity measurements, these studies provide us with an understanding of how much biological information can be accurately transferred between homologous proteins (71).
5.1 The bioinformatics spectrum
Figure 1 summarises the main points we raised in our discussions of organising and understanding biological data – the development of bioinformatics techniques has allowed an expansion of biological analysis in two dimension, depth and breadth. The first is represented by the vertical axis in the figure and outlines
a possible approach to the rational drug design process. The aim is to take a single protein and follow through an analysis that maximises our understanding of the protein it encodes. Starting with a gene sequence, we can determine the protein sequence with strong certainty. From there, prediction algorithms can be used to calculate the structure adopted by the protein. Geometry calculations can define the shape of the protein’s surface and molecular simulations can determine the force fields surrounding the molecule. Finally, using docking algorithms, one could identify or design ligands that may bind the protein, paving the way for designing a drug that specifically alters the protein’s function. In practise, the intermediate steps are still difficult to achieve accurately, and they are best combined with experimental methods to obtain some of the data, for example characterising the structure of the protein of interest.
The aims of the second dimension, the breadth in biological analysis, is to compare a gene with others. Initially, simple algorithms can be used to compare the sequences and structures of a pair of related proteins. With a larger number of proteins, improved algorithms can be used to produce multiple alignments, and extract sequence patterns or structural templates that define a family of proteins. Using this data, it is also possible to construct phylogenetic trees to trace the evolutionary path of proteins. Finally, with even more data, the information must be stored in large-scale databases. Comparisons become more complex, requiring multiple scoring schemes, and we are able to conduct genomic scale censuses that provide comprehensive statistical accounts of protein features, such as the abundance of particular structures or functions in different genomes. It also allows us to build phylogenetic trees that trace the evolution of whole organisms.
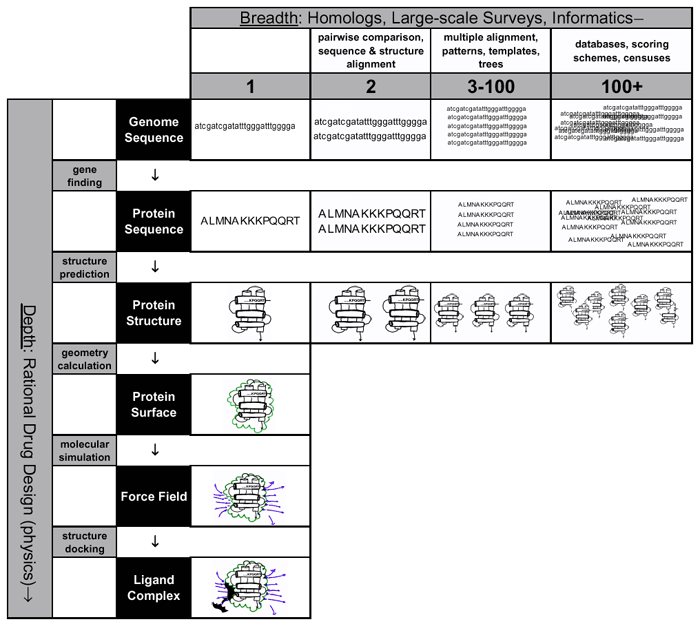
Figure
1. Paradigm shifts during the past
couple of decades have taken much of biology away from the laboratory bench and
have allowed the integration of other scientific disciplines, specifically
computing. The result is an expansion of biological research in breadth and
depth. The vertical axis demonstrates how bioinformatics can aid rational drug
design with minimal work in the wet lab. Starting with a single gene sequence,
we can determine with strong certainty, the protein sequence. From there, we
can determine the structure using structure prediction techniques. With
geometry calculations, we can further resolve the protein’s surface and through
molecular simulation determine the force fields surrounding the molecule.
Finally docking algorithms can provide predictions of the ligands that will
bind on the protein surface, thus paving the way for the design of a drug
specific to that molecule.
The horizontal axis shows how the influx of biological data and advances in computer technology have broadened the scope of biology. Initially with a pair of proteins, we can make comparisons between the between sequences and structures of evolutionary related proteins. With more data, algorithms for multiple alignments of several proteins become necessary. Using multiple sequences, we can also create phylogenetic trees to trace the evolutionary development of the proteins in question. Finally, with the deluge of data we currently face, we need to construct large databases to store, view and deconstruct the information. Alignments now become more complex, requiring sophisticated scoring schemes and there is enough data to compile a genome census – a genomic equivalent of a population census – providing comprehensive statistical accounting of protein features in genomes.
6. “… applying INFORMATICS TECHNIQUES…”
The distinct subject areas we mention require different types of informatics techniques. Briefly, for data organisation, the first biological databases were simple flat files. However with the increasing amount of information, relational database methods with Web-page interfaces have become increasingly popular. In sequence analysis, techniques include string comparison methods such as text search and 1D alignment algorithms. Motif and pattern identification for multiple sequences depend on machine learning, clustering and data-mining techniques. 3D structural analysis techniques include Euclidean geometry calculations combined with basic application of physical chemistry, graphical representations of surfaces and volumes, and structural comparison and 3D matching methods. For molecular simulations, Newtonian mechanics, quantum mechanics, molecular mechanics and electrostatic calculations are applied. In many of these areas, the computational methods must be combined with good statistical analyses in order to provide an objective measure for the significance of the results.
7. Transcription regulation – a case study in
bioinformatics
DNA-binding proteins have a central role in all aspects of genetic activity within an organism, participating in processes such as transcription, packaging, rearrangement, replication and repair. In this section, we focus on the studies that have contributed to our understanding of transcription regulation in different organisms. Through this example, we demonstrate how bioinformatics has been used to increase our knowledge of biological systems and also illustrate the practical applications of the different subject areas that were briefly outlined earlier. We start by considering structural analyses of how DNA-binding proteins recognise particular base sequences. Later, we review several genomic studies that have characterised the nature of transcription factors in different organisms, and the methods that have been used to identify regulatory binding sites in the upstream regions. Finally, we provide an overview of gene expression analyses that have been recently conducted and suggest future uses of transcription regulatory analyses to rationalise the observations made in gene expression experiments. All the results that we describe have been found through computational studies.
7.1 Structural studies
As of August 2000, there were 379 structures of protein-DNA complexes in the PDB. Analyses of these structures have provided valuable insight into the stereochemical principles of binding, including how particular base sequences are recognized and how the DNA structure is quite often modified on binding.
A structural taxonomy of DNA-binding proteins, similar to that presented in SCOP and CATH, was first proposed by Harrison (72) and periodically updated to accommodate new structures as they are solved (73). The classification consists of a two-tier system: the first level collects proteins into eight groups that share gross structural features for DNA-binding, and the second comprises 54 families of proteins that are structurally homologous to each other. Assembly of such a system simplifies the comparison of different binding methods; it highlights the diversity of protein-DNA complex geometries found in nature, but also underlines the importance of interactions between a-helices and the DNA major groove, the main mode of binding in over half the protein families. While the number of structures represented in the PDB does not necessarily reflect the relative importance of the different proteins in the cell, it is clear that helix-turn-helix, zinc-coordinating and leucine zipper motifs are used repeatedly. This provides compact frameworks that present the a-helix on the surfaces of structurally diverse proteins. At a gross level, it is possible to highlight the differences between transcription factor domains that “just” bind DNA and those involved in catalysis (74). Although there are exceptions, the former typically approach the DNA from a single face and slot into the grooves to interact with base edges. The latter commonly envelope the substrate, using complex networks of secondary structures and loops.
Focusing on proteins with a-helices, the structures show many variations, both in amino acid sequences and detailed geometry. They have clearly evolved independently in accordance with the requirements of the context in which they are found. While achieving a close fit between the a-helix and major groove, there is enough flexibility to allow both the protein and DNA to adopt distinct conformations. However, several studies that analysed the binding geometries of a-helices demonstrated that most adopt fairly uniform conformations regardless of protein family. They are commonly inserted in the major groove sideways, with their lengthwise axis roughly parallel to the slope outlined by the DNA backbone. Most start with the N-terminus in the groove and extend out, completing two to three turns within contacting distance of the nucleic acid (75, 76).
Given the similar binding orientations, it is surprising to find that the interactions between each amino acid position along the a-helices and nucleotides on the DNA vary considerably between different protein families. However, by classifying the amino acids according to the sizes of their side chains, we are able to rationalise the different interactions patterns. The rules of interactions are based on the simple premise that for a given residue position on a-helices in similar conformations, small amino acids interact with nucleotides that are close in distance and large amino acids with those that are further (76, 77). Equivalent studies for binding by other structural motifs, like b-hairpins, have also been conducted (78). When considering these interactions, it is important to remember that different regions of the protein surface also provide interfaces with the DNA.
This brings us to look at the atomic level interactions between individual amino acid-base pairs. Such analyses are based on the premise that a significant proportion of specific DNA-binding could be rationalised by a universal code of recognition between amino acids and bases, ie whether certain protein residues preferably interact with particular nucleotides regardless of the type of protein-DNA complex (79). Studies have considered hydrogen bonds, van der Waals contacts and water-mediated bonds (80-82). Results showed that about 2/3 of all interactions are with the DNA backbone and that their main role is one of sequence-independent stabilisation. In contrast, interactions with bases display some strong preferences, including the interactions of arginine or lysine with guanine, asparagine or glutamine with adenine and threonine with thymine. Such preferences were explained through examination of the stereochemistry of the amino acid side chains and base edges. Also highlighted were more complex types of interactions where single amino acids contact more than one base-step simultaneously, thus recognising a short DNA sequence. These results suggested that universal specificity, one that is observed across all protein-DNA complexes, indeed exists. However, many interactions that are normally considered to be non-specific, such as those with the DNA backbone, can also provide specificity depending on the context in which they are made.
Armed with an understanding of protein structure, DNA-binding motifs and side chain stereochemistry, a major application has been the prediction of binding either by proteins known to contain a particular motif, or those with structures solved in the uncomplexed form. Most common are predictions for a-helix-major groove interactions – given the amino acid sequence, what DNA sequence would it recognise (77, 83). In a different approach, molecular simulation techniques have been used to dock whole proteins and DNAs on the basis of force-field calculations around the two molecules (84, 85).
The reason that both methods have only been met with limited success is because even for apparently simple cases like a-helix-binding, there are many other factors that must be considered. Comparisons between bound and unbound nucleic acid structures show that DNA-bending is a common feature of complexes formed with transcription factors (74, 86). This and other factors such as electrostatic and cation-mediated interactions assist indirect recognition of the nucleotide sequence, although they are not well understood yet. Therefore, it is now clear that detailed rules for specific DNA-binding will be family specific, but with underlying trends such as the arginine-guanine interactions.
7.2 Genomic studies
Due to the wealth of biochemical data that are available, genomic studies in bioinformatics have concentrated on model organisms, and the analysis of regulatory systems has been no exception. Identification of transcription factors in genomes invariably depends on similarity search strategies, which assume a functional and evolutionary relationship between homologous proteins. In E. coli, studies have so far estimated a total of 300 to 500 transcription regulators (87) and PEDANT (88), a database of automatically assigned gene functions, shows that typically 2-3% of prokaryotic and 6-7% of eukaryotic genomes comprise DNA-binding proteins. As assignments were only complete for 40-60% of genomes as of August 2000, these figures most likely underestimate the actual number. Nonetheless, they already represent a large quantity of proteins and it is clear that there are more transcription regulators in eukaryotes than other species. This is unsurprising, considering the organisms have developed a relatively sophisticated transcription mechanism.
From the conclusions of the structural studies, the best strategy for characterising DNA-binding of the putative transcription factors in each genome is to group them by homology and analyse the individual families. Such classifications are provided in the secondary sequence databases described earlier and also those that specialise in regulatory proteins such as RegulonDB (89) and TRANSFAC (90). Of even greater use is the provision of structural assignments to the proteins; given a transcription factor, it is helpful to know the structural motif that it uses for binding, therefore providing us with a better understanding of how it recognises the target sequence. Structural genomics through bioinformatics assigns structures to the protein products of genomes by demonstrating similarity to proteins of known structure (91). These studies have shown that prokaryotic transcription factors most frequently contain helix-turn-helix motifs (87, 92) and eukaryotic factors contain homeodomain type helix-turn-helix, zinc finger or leucine zipper motifs. From the protein classifications in each genome, it is clear that different types of regulatory proteins differ in abundance and families significantly differ in size. A study by Huynen and van Nimwegen (93) has shown that members of a single family have similar functions, but as the requirements of this function vary over time, so does the presence of each gene family in the genome.
Most recently, using a combination of sequence and structural data, we examined the conservation of amino acid sequences between related DNA-binding proteins, and the effect that mutations have on DNA sequence recognition. The structural families described above were expanded to include proteins that are related by sequence similarity, but whose structures remain unsolved. Again, members of the same family are homologous, and probably derive from a common ancestor.
Amino acid conservations were calculated for the multiple sequence alignments of each family (94). Generally, alignment positions that interact with the DNA are better conserved than the rest of the protein surface, although the detailed patterns of conservation are quite complex. Residues that contact the DNA backbone are highly conserved in all protein families, providing a set of stabilising interactions that are common to all homologous proteins. The conservation of alignment positions that contact bases, and recognise the DNA sequence, are more complex and could be rationalised by defining a 3-class model for DNA-binding. First, protein families that bind non-specifically usually contain several conserved base-contacting residues; without exception, interactions are made in the minor groove where there is little discrimination between base types. The contacts are commonly used to stabilise deformations in the nucleic acid structure, particularly in widening the DNA minor groove. The second class comprise families whose members all target the same nucleotide sequence; here, base-contacting positions are absolutely or highly conserved allowing related proteins to target the same sequence.
The third, and most interesting, class comprises families in which binding is also specific but different members bind distinct base sequences. Here protein residues undergo frequent mutations, and family members can be divided into subfamilies according to the amino acid sequences at base-contacting positions; those in the same subfamily are predicted to bind the same DNA sequence and those of different subfamilies to bind distinct sequences. On the whole, the subfamilies corresponded well with the proteins’ functions and members of the same subfamilies were found to regulate similar transcription pathways. The combined analysis of sequence and structural data described by this study provided an insight into how homologous DNA-binding scaffolds achieve different specificities by altering their amino acid sequences. In doing so, proteins evolved distinct functions, therefore allowing structurally related transcription factors to regulate expression of different genes. Therefore, the relative abundance of transcription regulatory families in a genome depends, not only on the importance of a particular protein function, but also in the adaptability of the DNA-binding motifs to recognise distinct nucleotide sequences. This, in turn, appears to be best accommodated by simple binding motifs, such as the zinc fingers.
Given the knowledge of the transcription regulators that are contained in each organism, and an understanding of how they recognise DNA sequences, it is of interest to search for their potential binding sites within genome sequences (95). For prokaryotes, most analyses have involved compiling data on experimentally known binding sites for particular proteins and building a consensus sequence that incorporates any variations in nucleotides. Additional sites are found by conducting word-matching searches over the entire genome and scoring candidate sites by similarity (96-99). Unsurprisingly, most of the predicted sites are found in non-coding regions of the DNA (96) and the results of the studies are often presented in databases such as RegulonDB (89). The consensus search approach is often complemented by comparative genomic studies searching upstream regions of orthologous genes in closely related organisms. Through such an approach, it was found that at least 27% of known E. coli DNA-regulatory motifs are conserved in one or more distantly related bacteria (100).
The detection of regulatory sites in eukaryotes poses a more difficult problem because consensus sequences tend to be much shorter, variable, and dispersed over very large distances. However, initial studies in S. cerevisiae provided an interesting observation for the GATA protein in nitrogen metabolism regulation. While the 5 base-pair GATA consensus sequence is found almost everywhere in the genome, a single isolated binding site is insufficient to exert the regulatory function (101). Therefore specificity of GATA activity comes from the repetition of the consensus sequence within the upstream regions of controlled genes in multiple copies. An initial study has used this observation to predict new regulatory sites by searching for over-represented oligonucleotides in non-coding regions of yeast and worm genomes (102, 103).
Having detected the regulatory binding sites, there is the problem of defining the genes that are actually regulated, commonly termed regulons. Generally, binding sites are assumed to be located directly upstream of the regulons; however there are different problems associated with this assumption depending on the organism. For prokaryotes, it is complicated by the presence of operons; it is difficult to locate the regulated gene within an operon since it can lie several genes downstream of the regulatory sequence. It is often difficult to predict the organisation of operons (104), especially to define the gene that is found at the head, and there is often a lack of long-range conservation in gene order between related organisms (105). The problem in eukaryotes is even more severe; regulatory sites often act in both directions, binding sites are usually distant from regulons because of large intergenic regions, and transcription regulation is usually a result of combined action by multiple transcription factors in a combinatorial manner.
Despite these problems, these studies have succeeded in confirming the transcription regulatory pathways of well-characterised systems such as the heat shock response system (99). In addition, it is feasible to experimentally verify any predictions, most notably using gene expression data.
7.3 Gene expression studies
Many expression studies have so far focused on devising
methods to cluster genes by similarities in expression profiles. This is in
order to determine the proteins that are expressed together under different
cellular conditions. Briefly, the most common methods are hierarchical
clustering, self-organising maps, and K-means clustering. Hierarchical methods
originally derived from algorithms to construct phylogenetic trees, and group
genes in a “bottom-up” fashion; genes with the most similar expression profiles
are clustered first, and those with more diverse profiles are included
iteratively (106-108). In contrast, the self-organising map (109, 110) and K-means methods (111) employ a “top-down” approach in which the user
pre-defines the number of clusters for the dataset. The clusters are initially
assigned randomly, and the genes are regrouped iteratively until they are
optimally clustered.
Given these methods, it is
of interest to relate the expression data to other attributes such as
structure, function and subcellular localisation of each gene product. Mapping
these properties provide an insight into the characteristics of proteins that
are expressed together, and also suggest some interesting conclusions about the
overall biochemistry of the cell. In yeast, shorter proteins tend to be more
highly expressed than longer proteins, probably because of the relative ease
with which they are produced (112). Looking at the amino acid content, highly expressed
genes are generally enriched in alanine and glycine, and depleted in
asparagine; these are thought to reflect the requirements of amino acid usage
in the organism, where synthesis of alanine and glycine are energetically less
expensive than asparagine. Turning to protein structure, expression levels of
the TIM barrel and NTP hydrolase folds are highest, while those for the leucine
zipper, zinc finger and transmembrane helix-containing folds are lowest. This
relates to the functions associated with these folds; the former are commonly
involved in metabolic pathways and the latter in signalling or transport
processes (113). This is also reflected in the relationship with
subcellular localisations of proteins, where expression of cytoplasmic proteins
is high, but nuclear and membrane proteins tend to be low (114, 115).
More complex relationships
have also been assessed. Conventional wisdom is that gene products that
interact with each other are more likely to have similar expression profiles
than if they do not (116, 117). However, a recent study showed that this
relationship is not so simple (118). While expression profiles are similar for gene
products that are permanently associated, for example in the large ribosomal
subunit, profiles differ significantly for products that are only associated
transiently, including those belonging to the same metabolic pathway.
As described below, one of
the main driving forces behind expression analysis has been to analyse
cancerous cell lines (119). In general, it has been shown that different cell
lines (eg epithelial and ovarian cells) can be distinguished on the basis of
their expression profiles, and that these profiles are maintained when cells
are transferred from an in vivo to an in vitro environment (120). The basis for their physiological differences were
apparent in the expression of specific genes; for example, expression levels of
gene products necessary for progression through the cell cycle, especially
ribosomal genes, correlated well with variations in cell proliferation rate.
Comparative analysis can be extended to tumour cells, in which the underlying
causes of cancer can be uncovered by pinpointing areas of biological variations
compared to normal cells. For example in breast cancer, genes related to cell
proliferation and the IFN-regulated signal transduction pathway were found to
be upregulated (52, 121). One of the difficulties in cancer treatment has been
to target specific therapies to pathogenetically distinct tumour types, in
order to maximise efficacy and minimise toxicity. Therefore, improvements in
cancer classifications have been central to advances in cancer treatment.
Although the distinction between different forms of cancer – for example subclasses
of acute leukaemia – has been well established, it is still not possible to
establish a clinical diagnosis on the basis of a single test. In a recent study, acute myeloid leukaemia
and acute lymphoblastic leukaemia were successfully distinguished based on the
expression profiles of these cells (53). As the approach does not require prior biological
knowledge of the diseases, it may provide a generic strategy for classifying
all types of cancer.
Clearly, an essential aspect of understanding expression data lies in understanding the basis of transcription regulation. However, analysis in this area is still limited to preliminary analyses of expression levels in yeast mutants lacking key components of the transcription initiation complex (10, 122).
8. “… many PRACTICAL APPLICATIONS…”
Here, we describe some of the major uses of bioinformatics.
8.1 Finding Homologues
As described earlier, one of the driving forces behind bioinformatics is the search for similarities between different biomolecules. Apart from enabling systematic organisation of data, identification of protein homologues has some direct practical uses. The most obvious is transferring information between related proteins. For example, given a poorly characterised protein, it is possible to search for homologues that are better understood and with caution, apply some of the knowledge of the latter to the former. Specifically with structural data, theoretical models of proteins are usually based on experimentally solved structures of close homologues (123). Similar techniques are used in fold recognition in which tertiary structure predictions depend on finding structures of remote homologues and checking whether the prediction is energetically viable (124). Where biochemical or structural data are lacking, studies could be made in low-level organisms like yeast and the results applied to homologues in higher-level organisms such as humans, where experiments are more demanding.
An equivalent approach is also employed in genomics. Homologue-finding is extensively used to confirm coding regions in newly sequenced genomes and functional data is frequently transferred to annotate individual genes. On a larger scale, it also simplifies the problem of understanding complex genomes by analysing simple organisms first and then applying the same principles to more complicated ones – this is one reason why early structural genomics projects focused on Mycoplasma genitalium (91).
Ironically, the same idea can be applied in reverse. Potential drug targets are quickly discovered by checking whether homologues of essential microbial proteins are missing in humans. On a smaller scale, structural differences between similar proteins may be harnessed to design drug molecules that specifically bind to one structure but not another.
8.2 Rational Drug Design
One
of the earliest medical applications of bioinformatics has been in aiding
rational drug design. Figure 2 outlines the commonly cited approach, taking the
MLH1 gene product as an example drug target. MLH1 is a human gene encoding a mismatch repair protein (mmr) situated
on the short arm of chromosome 3 (125). Through linkage analysis and its similarity to mmr
genes in mice, the gene has been implicated in nonpolyposis colorectal cancer (126). Given the nucleotide sequence, the probable amino
acid sequence of the encoded protein can be determined using translation
software. Sequence search techniques can then be used to find homologues in
model organisms, and based on sequence similarity, it is possible to model the
structure of the human protein on experimentally characterised structures.
Finally, docking algorithms could design molecules that could bind the model
structure, leading the way for biochemical assays to test their biological
activity on the actual protein.
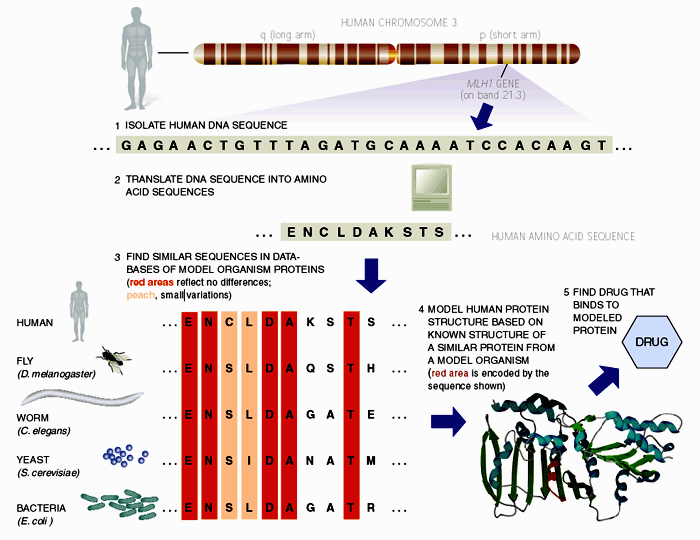
Figure 2. Above is a schematic outlining how scientists can use bioinformatics to aid rational drug discovery. MLH1 is a human gene encoding a mismatch repair protein (mmr) situated on the short arm of chromosome 3. Through linkage analysis and its similarity to mmr genes in mice, the gene has been implicated in nonpolyposis colorectal cancer. Given the nucleotide sequence, the probable amino acid sequence of the encoded protein can be determined using translation software. Sequence search techniques can be used to find homologues in model organisms, and based on sequence similarity, it is possible to model the structure of the human protein on experimentally characterised structures. Finally, docking algorithms could design molecules that could bind the model structure, leading the way for biochemical assays to test their biological activity on the actual protein.
8.3 Large-scale censuses
Although databases can efficiently store all the information related to genomes, structures and expression datasets, it is useful to condense all this information into understandable trends and facts that users can readily understand. Broad generalisations help identify interesting subject areas for further detailed analysis, and place new observations in a proper context. This enables one to see whether they are unusual in any way.
Through these large-scale censuses, one can address a number of evolutionary, biochemical and biophysical questions. For example, are specific protein folds associated with certain phylogenetic groups? How common are different folds within particular organisms? And to what degree are folds shared between related organisms? Does this extent of sharing parallel measures of relatedness derived from traditional evolutionary trees? Initial studies show that the frequency of folds differs greatly between organisms and that the sharing of folds between organisms does in fact follow traditional phylogenetic classifications (21, 41). We can also integrate data on protein functions; given that the particular protein folds are often related to specific biochemical functions (68, 69), these findings highlight the diversity of metabolic pathways in different organisms (20, 105).
As we discussed earlier, one of the most exciting new sources of genomic information is the expression data. Combining expression information with structural and functional classifications of proteins we can ask whether the high occurrence of a protein fold in a genome is indicative of high expression levels (112). Further genomic scale data that we can consider in large-scale surveys include the subcellular localisations of proteins and their interactions with each other (127-129). In conjunction with structural data, we can then begin to compile a map of all protein-protein interactions in an organism.
8.4 Further applications in medical sciences
Most recent applications in the medical sciences have centred on gene expression analysis (130). This usually involves compiling expression data for cells affected by different diseases (131), eg cancer (53, 132, 133) and ateriosclerosis (134), and comparing the measurements against normal expression levels. Identification of genes that are expressed differently in affected cells provides a basis for explaining the causes of illnesses and highlights potential drug targets. Using the process described in Figure 2, one would design compounds that bind the expressed protein, or perhaps more importantly, the transcription regulator has caused the change in expression levels. Given a lead compound, microarray experiments can then be used to evaluate responses to pharmacological intervention, (135, 136) and also provide early tests to detect or predict the toxicity of trial drugs.
Further advances in bioinformatics combined with experimental genomics for individuals are predicted to revolutionalise the future of healthcare. A typical scenario for a patient may start with post-natal genotyping to assess susceptibility or immunity from specific diseases and pathogens. With this information, a unique combination of vaccines could be prescribed, minimising the healthcare costs of unnecessary treatments and anticipating the onslaught of diseases later in life. Regular lifetime screenings could lead to guidance for nutrition intake and early detections of any illnesses (137). In addition, drug-based treatments could be tailored specifically to the patient and disease, thus providing the most effective course of medication with minimal side-effects (138). Given the present rate of development, such a scenario in healthcare appears to be possible in the not too distant future.
9. Conclusions
With the current deluge of data, computational methods have become indispensable to biological investigations. Originally developed for the analysis of biological sequences, bioinformatics now encompasses a wide range of subject areas including structural biology, genomics and gene expression studies. In this review, we provided an introduction and overview of the current state of field. In particular, we discussed the types of biological information and databases that are commonly used, examined some of the studies that are being conducted – with reference to transcription regulatory systems – and finally looked at several practical applications of the field.
Two principal approaches underpin all studies in bioinformatics. First is that of comparing and grouping the data according to biologically meaningful similarities and second, that of analysing one type of data to infer and understand the observations for another type of data. These approaches are reflected in the main aims of the field, which are to understand and organise the information associated with biological molecules on a large scale. As a result, bioinformatics has not only provided greater depth to biological investigations, but added the dimension of breadth as well. In this way, we are able to examine individual systems in detail and also compare them with those that are related in order to uncover common principles that apply across many systems and highlight unusual features that are unique to some.
10. Acknowledgements
We thank Patrick McGarvey for comments on the manuscript.
11. References
1. Reichhardt T. It's sink or swim as a tidal wave of data approaches. Nature 1999;399(6736):517-20.
2. Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Rapp BA, Wheeler DL. GenBank. Nucleic Acids Res 2000;28(1):15-8.
3. Bairoch A, Apweiler R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res 2000;28(1):45-8.
4. Fleischmann RD, Adams MD, White O, Clayton RA, Kirkness EF, Kerlavage AR, et al. Whole-genome random sequencing and assembly of Haemophilus influenzae. Science 1995;269(5223):496-512.
5. Drowning in data. The Economist 26 June 1999.
6. Bernstein FC, Koetzle TF, Williams GJ, Meyer EF, Jr., Brice MD, Rodgers JR, et al. The Protein Data Bank. A computer-based archival file for macromolecular structures. Eur J Biochem 1977;80(2):319-24.
7. Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. The Protein Data Bank. Nucleic Acids Res 2000;28(1):235-42.
8. Pearson WR, Lipman DJ. Improved tools for biological sequence comparison. Proc Natl Acad Sci U S A 1988;85(8):2444-2448.
9. Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389-3402.
10. Holstege FC JE, Wyrick JJ, Lee TI, Hengartner CJ, Green MR, Golub TR, Lander ES, Young RA. Dissecting the regulatory circuitry of a eukaryotic genome. Cell 1998;95(5):717-728.
11. Pedersendagger AG, Jensendagger LJ, Brunak S, Staerfeldt HH, Ussery DW. A DNA structural atlas for Escherichia coli. J Mol Biol 2000;299(4):907-930.
12. Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 2000;28(1):27-30.
13. Jeffery CJ. Moonlighting proteins. TIBS 1999;24(1):8-11.
14. Chothia C. Proteins. One thousand families for the molecular biologist. Nature 1992;357(6379):543-4.
15. Orengo CA, Jones DT, Thornton JM. Protein superfamilies and domain superfolds. Nature 1994;372(6507):631-4.
16. Lesk AM, Chothia C. How different amino acid sequences determine similar protein structures: the structure and evolutionary dynamics of the globins. J Mol Biol 1980;136(3):225-70.
17. Russell RB, Saqi MA, Sayle RA, Bates PA, Sternberg MJ. Recognition of analogous and homologous protein folds: analysis of sequence and structure conservation. J Mol Biol 1997;269(3):423-39.
18. Russell RB, Saqi MA, Bates PA, Sayle RA, Sternberg MJ. Recognition of analogous and homologous protein folds--assessment of prediction success and associated alignment accuracy using empirical substitution matrices. Protein Eng 1998;11(1):1-9.
19. Fitch WM. Distinguishing homologous from analogous proteins. Syst Zool 1970;19:99-110.
20. Tatusov RL, Koonin EV, Lipman DJ. A genomic perspective on protein families. Science 1997;278(5338):631-7.
21. Gerstein M, Hegyi H. Comparing genomes in terms of protein structure: surveys of a finite parts list. FEMS Microbiol Rev 1998;22(4):277-304.
22. Skolnick J, Fetrow JS. From genes to protein structure and function: novel applications of computational approaches in the genomic era. TIBtech 2000;18:34-39.
23. McGarvey PB, Huang H, Barker WC, Orcutt BC, Garavelli JS, Srinivasarao GY, et al. PIR: a new resource for bioinformatics. Bioinformatics 2000;16(3):290-291.
24. Bleasby AJ, Akrigg D, Attwood TK. OWL--a non-redundant composite protein sequence database. Nucleic Acids Res 1994;22(17):3574-3577.
25. Bleasby AJ, Wootton JC. Construction of validated, non-redundant composite protein sequence databases. Protein Eng 1990;3(3):153-159.
26. Hofmann K, Bucher P, Falquet L, Bairoch A. The PROSITE database, its status in 1999. Nucleic Acids Res 1999;27(1):215-219.
27. Attwood TK, Croning MD, Flower DR, Lewis AP, Mabey JE, Scordis P, et al. PRINTS-S: the database formerly known as PRINTS. Nucleic Acids Res 2000;28(1):225-227.
28. Bateman A, Birney E, Durbin R, Eddy SR, Howe KL, Sonnhammer EL. The Pfam protein families database. Nucleic Acids Res 2000;28(1):263-266.
29. Attwood TK, Flower DR, Lewis AP, Mabey JE, Morgan SR, Scordis P, et al. PRINTS prepares for the new millennium. Nucleic Acids Res 1999;27(1):220-225.
30. Laskowski RA, Hutchinson EG, Michie AD, Wallace AC, Jones ML, Thornton JM. PDBsum: a Web-based database of summaries and analyses of all PDB structures. TIBS 1997;22(12):488-490.
31. Pearl FM, Lee D, Bray JE, Sillitoe I, Todd AE, Harrison AP, et al. Assigning genomic sequences to CATH. Nucleic Acids Res 2000;28(1):277-282.
32. Lo Conte L, Ailey B, Hubbard TJ, Brenner SE, Murzin AG, Chothia C. SCOP: a structural classification of proteins database. Nucleic Acids Res 2000;28(1):257-259.
33. Holm L, Sander C. Touring protein fold space with Dali/FSSP. Nucleic Acids Res 1998;26(1):316-319.
34. Berman HM, Olson WK, Beveridge DL, Westbrook J, Gelbin A, Demeny T, et al. The Nucleic Acid Database. A comprehensive relational database of three-dimensional structures of nucleic acids. Biophys J 1992;63(3):751-759.
35. Vondrasek J, Wlodawer A. Database of HIV proteinase structures. TIBS 1997;22(5):183.
36. Hendlich M. Databases for protein-ligand complexes. Acta Cryst D 1998;54(1):1178-1182.
37. Baker W, van den Broek A, Camon E, Hingamp P, Sterk P, Stoesser G, et al. The EMBL nucleotide sequence database. Nucleic Acids Res 2000;28(1):19-23.
38. Okayama T, Tamura T, Gojobori T, Tateno Y, Ikeo K, Miyazaki S, et al. Formal design and implementation of an improved DDBJ DNA database with a new schema and object-oriented library. Bioinformatics 1998;14(6):472-8.
39. Schuler GD, Epstein JA, Ohkawa H, Kans JA. Entrez: molecular biology database and retrieval system. Methods Enzymol 1996;266:141-62.
40. Tatusova TA, Karsch-Mizrachi I, Ostell JA. Complete genomes in WWW Entrez: data representation and analysis. Bioinformatics 1999;15(7-8):536-43.
41. Lin J, Gerstein M. Whole-genome trees based on the occurrence of folds and orthologs: implications for comparing genomes on different levels. Genome Res 2000;10(6):808-18.
42. Eisen MB, Brown PO. DNA arrays for analysis of gene expression. Methods Enzymol 1999;303:179-205.
43. Cheung VG, Morley M, Aguilar F, Massimi A, Kucherlapati R, Childs G. Making and reading microarrays. Nat Genet 1999;21(1 Suppl):15-9.
44. Duggan DJ, Bittner M, Chen Y, Meltzer P, Trent JM. Expression profiling using cDNA microarrays. Nat Genet 1999;21(1 Suppl):10-4.
45. Lipshutz RJ FS, Gingeras TR, Lockhart DJ. High density synthetic oligonucleotide arrays. Nat Gen 1999;21(1):20-24.
46. Velculescu VE ZL, Zhou, W Traverso, G St Croix, B Vogelstein B, Kinzler KW. Serial Analysis of Gene Expression Detailed Protocol. 1999.
47. Roth FP HJ, Estep PW, Church GM. Finding DNA regulatory motifs within unaligned noncoding sequences clustered by whole-genome mRNA quantitation. Nat Biotechnol 1998;16(10):939-45.
48. Jelinsky SA, Samson LD. Global response of Saccharomyces cerevisiae to an alkylating agent. Proc Natl Acad Sci U S A 1999;96(4):1486-91.
49. Cho RJ, Campbell MJ, Winzeler EA, Steinmetz L, Conway A, Wodicka L, et al. A genome-wide transcriptional analysis of the mitotic cell cycle. Mol Cell 1998;2(1):65-73.
50. DeRisi JL, Iyer VR, Brown PO. Exploring the metabolic and genetic control of gene expression on a genomic scale. Science 1997;278(5338):680-6.
51. Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, Andre B, et al. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 1999;285(5429):901-6.
52. Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, et al. Molecular portraits of human breast tumours. Nature 2000;406(6797):747-52.
53. Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science 1999;286(5439):531-7.
54. Celis JE, Gromov P. 2D protein electrophoresis: can it be perfected? Curr Opin Biotechnol 1999;10(1):16-21.
55. Pandey A, Mann M. Proteomics to study genes and genomes. Nature 2000;405(6788):837-46.
56. Futcher B, Latter GI, Monardo P, McLaughlin CS, Garrels JI. A sampling of the yeast proteome. Mol Cell Biol 1999;19(11):7357-68.
57. Gygi SP, Rist B, Gerber SA, Turecek F, Gelb MH, Aebersold R. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat Biotechnol 1999;17(10):994-9.
58. Gerstein M. Integrative database analysis in structural genomics. Nature Struct Biol In Press.
59. Etzold T, Ulyanov A, Argos P. SRS: information retrieval system for molecular biology data banks. Methods Enzymol 1996;266:114-28.
60. Wade K. Searching Entrez PubMed and uncover on the internet. Aviat Space Environ Med 2000;71(5):559.
61. Zhang MQ. Promoter analysis of co-regulated genes in the yeast genome. Comput Chem 1999;23(3-4):233-50.
62. Boguski MS. Biosequence exegesis. Science 1999;286(5439):453-5.
63. Miller C, Gurd J, Brass A. A RAPID algorithm for sequence database comparisons: application to the identification of vector contamination in the EMBL databases. Bioinformatics 1999;15(2):111-21.
64. Gonnet GH, Korostensky C, Benner S. Evaluation measures of multiple sequence alignments. J Comput Biol 2000;7(1-2):261-76.
65. Orengo CA, Taylor WR. SSAP: sequential structure alignment program for protein structure comparison. Methods Enzymol 1996;266:617-35.
66. Orengo CA. CORA--topological fingerprints for protein structural families. Protein Sci 1999;8(4):699-715.
67. Russell RB, Sternberg MJ. Structure prediction. How good are we? Curr Biol 1995;5(5):488-90.
68. Martin AC, Orengo CA, Hutchinson EG, Jones S, Karmirantzou M, Laskowski RA, et al. Protein folds and functions. Structure 1998;6(7):875-84.
69. Hegyi H, Gerstein M. The relationship between protein structure and function: a comprehensive survey with application to the yeast genome. J Mol Biol 1999;288(1):147-64.
70. Russell RB, Sasieni PD, Sternberg MJE. Supersites within superfolds. Binding site similarity in the absence of homology. J Mol Biol 1998;282(4):903-18.
71. Wilson CA, Kreychman J, Gerstein M. Assessing annotation transfer for genomics: quantifying the relations between protein sequence, structure and function through traditional and probabilistic scores. J Mol Biol 2000;297(1):233-49.
72. Harrison SC. A structural taxonomy of DNA-binding domains. Nature 1991;353(6346):715-9.
73. Luscombe NM, Austin SE, Berman HM, Thornton JM. An overview of the structures of protein-DNA complexes. Genome Biology 2000;1(1):1-37.
74. Jones S, van Heyningen P, Berman HM, Thornton JM. Protein-DNA interactions: A structural analysis. J Mol Biol 1999;287(5):877-96.
75. Suzuki M, Gerstein M. Binding geometry of alpha-helices that recognize DNA. Proteins 1995;23(4):525-35.
76. Luscombe NM, Thornton JM. Protein-DNA interactions: a 3D analysis of alpha-helix-binding in the major groove. Manuscript in preparation.
77. Suzuki M, Brenner SE, Gerstein M, Yagi N. DNA recognition code of transcription factors. Protein Eng 1995;8(4):319-28.
78. Suzuki M. DNA recognition by a beta-sheet. Protein Eng 1995;8(1):1-4.
79. Seeman NC, Rosenberg JM, Rich A. Sequence specific recognition of double helical nucleic acids by proteins. Proc Natl Acad Sci U S A 1976;73:804-808.
80. Suzuki M. A framework for the DNA-protein recognition code of the probe helix in transcription factors: the chemical and stereochemical rules. Structure 1994;2(4):317-26.
81. Mandel-Gutfreund Y, Schueler O, Margalit H. Comprehensive analysis of hydrogen bonds in regulatory protein DNA- complexes: in search of common principles. J Mol Biol 1995;253(2):370-82.
82. Luscombe NM, Laskowski RA, Thornton JM. Protein-DNA interactions: a 3D analysis of amino acid-base interactions. Manuscript in preparation.
83. Mandel-Gutfreund Y, Margalit H, Jernigan RL, Zhurkin VB. A role for CH...O interactions in protein-DNA recognition. J Mol Biol 1998;277(5):1129-40.
84. Sternberg MJ, Gabb HA, Jackson RM. Predictive docking of protein-protein and protein-DNA complexes. Curr Opin Struct Biol 1998;8(2):250-6.
85. Aloy P, Moont G, Gabb HA, Querol E, Aviles FX, Sternberg MJ. Modelling repressor proteins docking to DNA. Proteins 1998;33(4):535-49.
86. Dickerson RE. DNA bending: the prevalence of kinkiness and the virtues of normality. Nucleic Acids Res 1998;26(8):1906-26.
87. Perez-Rueda E, Collado-Vides J. The repertoire of DNA-binding transcriptional regulators in Escherichia coli K-12. Nucleic Acids Res 2000;28(8):1838-47.
88. Mewes HW, Frishman D, Gruber C, Geier B, Haase D, Kaps A, et al. MIPS: a database for genomes and protein sequences. Nucleic Acids Res 2000;28(1):37-40.
89. Salgado H, Santos-Zavaleta A, Gama-Castro S, Millan-Zarate D, Blattner FR, Collado-Vides J. RegulonDB (version 3.0): transcriptional regulation and operon organization in Escherichia coli K-12. Nucleic Acids Res 2000;28(1):65-7.
90. Wingender E, Chen X, Hehl R, Karas H, Liebich I, Matys V, et al. TRANSFAC: an integrated system for gene expression regulation. Nucleic Acids Res 2000;28(1):316-9.
91. Teichmann SA, Chothia C, Gerstein M. Advances in structural genomics. Curr Opin Struct Biol 1999;9(3):390-9.
92. Aravind L, Koonin EV. DNA-binding proteins and evolution of transcription regulation in the archaea. Nucleic Acids Res 1999;27(23):4658-70.
93. Huynen MA, van Nimwegen E. The frequency distribution of gene family sizes in complete genomes. Mol Biol Evol 1998;15(5):583-9.
94. Luscombe NM, Thornton JM. Protein-DNA interactions: an analysis of amino acid conservation and the effect on binding specificity. Manuscript in preparation.
95. Gelfand MS. Prediction of function in DNA sequence analysis. J Comp Biol 1995;1:87-115.
96. Robison K, McGuire AM, Church GM. A comprehensive library of DNA-binding site matrices for 55 proteins applied to the complete Escherichia coli K-12 genome. J Mol Biol 1998;284(2):241-54.
97. Thieffry D, Salgado H, Huerta AM, Collado-Vides J. Prediction of transcriptional regulatory sites in the complete genome sequence of Escherichia coli K-12. Bioinformatics 1998;14(5):391-400.
98. Mironov AA, Koonin EV, Roytberg MA, Gelfand MS. Computer analysis of transcription regulatory patterns in completely sequenced bacterial genomes. Nucleic Acids Res 1999;27(14):2981-9.
99. Gelfand MS, Koonin EV, Mironov AA. Prediction of transcription regulatory sites in Archaea by a comparative genomic approach. Nucleic Acids Res 2000;28(3):695-705.
100. McGuire AM, Hughes JD, Church GM. Conservation of DNA regulatory motifs and discovery of new motifs in microbial genomes. Genome Res 2000;10(6):744-57.
101. Bysani N, Daugherty JR, Cooper TG. Saturation mutagenesis of the UASNTR (GATAA) responsible for nitrogen catabolite repression-sensitive transcriptional activation of the allantoin pathway genes in Saccharomyces cerevisiae. J Bacteriol 1991;173(16):4977-82.
102. Clarke ND, Berg JM. Zinc fingers in Caenorhabditis elegans: finding families and probing pathways. Science 1998;282(5396):2018-22.
103. van Helden J, Andre B, Collado-Vides J. Extracting regulatory sites from the upstream region of yeast genes by computational analysis of oligonucleotide frequencies. J Mol Biol 1998;281(5):827-42.
104. Salgado H, Moreno-Hagelsieb G, Smith TF, Collado-Vides J. Operons in Escherichia coli: genomic analyses and predictions. Proc Natl Acad Sci U S A 2000;97(12):6652-7.
105. Tatusov RL, Mushegian AR, Bork P, Brown NP, Hayes WS, Borodovsky M, et al. Metabolism and evolution of Haemophilus influenzae deduced from a whole- genome comparison with Escherichia coli. Curr Biol 1996;6(3):279-91.
106. Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A 1998;95(25):14863-8.
107. Wen X, Fuhrman S, Michaels GS, Carr DB, Smith S, Barker JL, et al. Large-scale temporal gene expression mapping of central nervous system development. Proc Natl Acad Sci U S A 1998;95(1):334-9.
108. Alon U, Barkai N, Notterman DA, Gish K, Ybarra S, Mack D, et al. Broad patterns of gene expression revealed by clustering analysis of tumor and normal colon tissues probed by oligonucleotide arrays. Proc Natl Acad Sci U S A 1999;96(12):6745-50.
109. Tamayo P, Slonim D, Mesirov J, Zhu Q, Kitareewan S, Dmitrovsky E, et al. Interpreting patterns of gene expression with self-organizing maps: methods and application to hematopoietic differentiation. Proc Natl Acad Sci U S A 1999;96(6):2907-12.
110. Toronen P, Kolehmainen M, Wong G, Castren E. Analysis of gene expression data using self-organizing maps. FEBS Lett 1999;451(2):142-6.
111. Tavazoie S, Hughes JD, Campbell MJ, Cho RJ, Church GM. Systematic determination of genetic network architecture. Nat Genet 1999;22(3):281-5.
112. Jansen R, Gerstein M. Analysis of the yeast transcriptome with structural and functional categories: characterizing highly expressed proteins. Nucleic Acids Res 2000;28(6):1481-8.
113. Gerstein M, Jansen R. The current excitment in bioinformatics, analysis of whole-genome expression data: how does it relate to protein structure and function. Current Opinion in Structural Biology In Press.
114. Drawid A, Gerstein M. A Bayesian System Integrating Expression Data with Sequence Patterns for Localizing Proteins: Comprehensive Application to the Yeast Genome. J Mol Biol In press.
115. Drawid A, Jansen R, Gerstein M. Genom-wide analysis relating expression level with protein subcellular localisation. TIGS In press.
116. Marcotte EM, Pellegrini M, Ng HL, Rice DW, Yeates TO, Eisenberg D. Detecting protein function and protein-protein interactions from genome sequences. Science 1999;285(5428):751-3.
117. Eisenberg D, Marcotte EM, Xenarios I, Yeates TO. Protein function in the post-genomic era. Nature 2000;405(6788):823-6.
118. Jansen R, Greenbaum D, Gerstein M. Relating whole-genome expression data with protein-protein interactions. Manuscript in preparation.
119. Marx J. Medicine. DNA arrays reveal cancer in its many forms. Science 2000;289(5485):1670-2.
120. Ross DT, Scherf U, Eisen MB, Perou CM, Rees C, Spellman P, et al. Systematic variation in gene expression patterns in human cancer cell lines. Nat Genet 2000;24(3):227-35.
121. Perou CM, Jeffrey SS, van de Rijn M, Rees CA, Eisen MB, Ross DT, et al. Distinctive gene expression patterns in human mammary epithelial cells and breast cancers. Proc Natl Acad Sci U S A 1999;96(16):9212-7.
122. Livesey FJ, Furukawa T, Steffen MA, Church GM, Cepko CL. Microarray analysis of the transcriptional network controlled by the photoreceptor homeobox gene Crx. Curr Biol 2000;10(6):301-10.
123. Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 1993;234(3):779-815.
124. Jones DT, Taylor WR, Thornton JM. A new approach to protein fold recognition. Nature 1992;358(6381):86-9.
125. Kok K, Naylor SL, Buys CH. Deletions of the short arm of chromosome 3 in solid tumors and the search for suppressor genes. Adv Cancer Res 1997;71:27-92.
126. Syngal S, Fox EA, Eng C, Kolodner RD, Garber JE. Sensitivity and specificity of clinical criteria for hereditary non-polyposis colorectal cancer associated mutations in MSH2 and MLH1. J Med Gen 2000;37(9):641-645.
127. Uetz P, Giot L, Cagney G, Mansfield TA, Judson RS, Knight JR, et al. A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature 2000;403(6770):623-7.
128. Ross-Macdonald P, Sheehan A, Friddle C, Roeder GS, Snyder M. Transposon mutagenesis for the analysis of protein production, function, and localization. Methods Enzymol 1999;303:512-32.
129. Mewes HW, Heumann K, Kaps A, Mayer K, Pfeiffer F, Stocker S, et al. MIPS: a database for genomes and protein sequences. Nucleic Acids Res 1999;27(1):44-8.
130. Murray-Rust P. Bioinformatics and drug discovery. Curr Opin Biotechnol 1994;5(6):648-53.
131. Friend SH. How DNA microarrays and expression profiling will affect clinical practice. BMJ 1999;319(7220):1306-7.
132. Tamayo P SD, Mesirov J, Zhu Q, Kitareewan S, Dmitrovsky E, Lander ES, Golub TR. Interpreting patterns of gene expression with self-organizing maps: methods and application to hematopoietic differentiation. Proc Natl Acad Sci U S A 1999;96(6):2907-12.
133. Perou CM JS, van de Rijn M, Rees CA, Eisen MB, Ross DT, Pergamenschikov A, Williams CF, Zhu SX, Lee JC, Lashkari D, Shalon D, Brown PO, Botstein D. Distinctive gene expression patterns in human mammary epithelial cells and breast cancers. Proc Natl Acad Sci 1999;96(16):9212-7.
134. Hiltunen MO, Niemi M, Yla-Herttuala S. Functional genomics and DNA array techniques in atherosclerosis research. Curr Opin Lipidol 1999;10(6):515-9.
135. Colantuoni C, Purcell AE, Bouton CM, Pevsner J. High throughput analysis of gene expression in the human brain. J Neurosci Res 2000;59(1):1-10.
136. Debouck C, Metcalf B. The impact of genomics on drug discovery. Annu Rev Pharmacol Toxicol 2000;40:193-207.
137. Sander C. Genomic medicine and the future of health care. Science 2000;287(5460):1977-8.
138. Ohlstein EH, Ruffolo RR, Jr., Elliott JD. Drug discovery in the next millennium. Annu Rev Pharmacol Toxicol 2000;40:177-91.